En la anterior publicación del SAI de Resonancia Magnética de Investigación nos centramos en los agentes de contraste que producían un contraste positivo, concretamente en los agentes de Gadolinio. En esta nueva entrada, nos gustaría hablar de cómo podemos obtener un contraste positivo empleando partículas de óxido de hierro, ya que hasta ahora, estas partículas las incluíamos como una fuente de contraste negativo. Todo dependerá de ciertas propiedades fisicoquímicas: tamaño, morfología y superficie de las partículas.
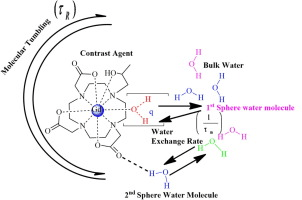
Las partículas de óxido de hierro son uno de los nanomateriales más usados en biomedicina, y dos de sus propiedades más importantes son su biocompatibilidad y sus propiedades magnéticas.
Cuando son sintetizadas de manera “tradicional” muestran propiedades superparamagnéticas, cuando se les somete a un campo magnético muestran una fuerte respuesta magnética, pero vuelve a cero cuando se elimina el campo. Es decir, actúan como “pequeños imanes” eliminando la señal, y apareciendo un “punto negro” produciendo lo que se llama contraste negativo. Como su efecto es bastante acusado, para aplicaciones in vivo la dosis administrada suele ser baja.
Si os acordáis de la anterior newsletter, hablamos del contraste de Gd, que es el que se utiliza la mayoría de las veces, pero se ha visto que produce toxicidad con lo que las investigaciones se centran en estudiar nuevos compuestos que puedan producir contraste positivo empleando otros elementos.
Con estas partículas de hierro, el contraste negativo produce imágenes oscuras. En la mayoría de las enfermedades, la señal que se produce en la imagen de RM es oscura. Por tanto, si el contraste produce una señal oscura sobre un fondo oscuro, el diagnóstico va a ser complicado, de ahí que, en estos años, los investigadores estén buscando alternativas para mejorar las propiedades fisicoquímicas de las partículas de óxido de hierro para conseguir que las imágenes que se obtengan produzcan el mismo contraste que el que se produce cuando se emplean compuestos de Gadolinio.
La intención será sintetizar partículas muy pequeñas que sean más paramagnéticas que superparamagnéticas y que produzcan señales hiperintensas en imagen por RM.
Propiedades fisicoquímicas de las partículas de óxido de hierro
Para conseguir que las partículas de óxido de hierro actúen como agentes de T1: Los componentes de estas partículas: el núcleo y la envoltura desempeñarán un papel muy importante en el contraste. La composición, la forma y el tamaño son clave para determinar la fase de hierro-oxido, su cristalinidad, las propiedades magnéticas y su tamaño hidrodinámico.
Con respecto al tamaño, (considerando tamaño del núcleo y tamaño hidrodinámico): cuando más pequeño sea el núcleo mejor será el contraste positivo. Cuando el tamaño del núcleo es menor a <5nm los dominios magnéticos individuales disminuyen, produciendo un giro inclinado que produce 5 electrones orientados y desapareados Fe3+/Fe2+, haciendo a las partículas más paramagnéticas que superparamagnéticas. Los núcleos más pequeños tienen menos cristalinidad, con lo que se produce una disminución en la saturación de la magnetización (similar a los materiales paramagnéticos).
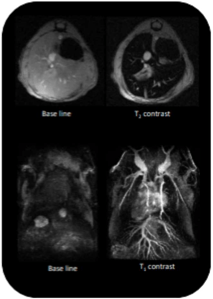
Imagen de hígado ponderada en T1: basal (izda) y después de administrar nanopartículas de hierro con un tamaño relativamente grande (dcha.) Se observa una hipointensidad en la señal. Angiografía en T1: antes de administrar el contraste (izda) y después de administrar nanopartículas de hierro con un tamaño muy inferior a las anteriores. Se produce una hiperintensidad en la señal.
Un aspecto muy importante que hay que tener en cuenta es la estabilidad de estas NPs (nanopartículas) para evitar que se agreguen entre ellas, pues esto produciría un aumento en el efecto T2. Serán cruciales una serie de aspectos durante su síntesis, ya no solo el tamaño en si, si no la envoltura, el tipo y tamaño, la hidrodinámica, etc… no hay una regla general que defina el mejor escenario para cada nanopartícula de T1. Hay otros factores, como la secuencia en sí de T1, la elección de los parámetros en diferentes campos magnéticos para que se puedan emplear estas NPS con unas propiedades fisicoquímicas y relaxométricas concretas.
Aplicaciones in vivo
Como ya comentamos, el principal uso de estas NPS de hierro es para servir de contraste T2, pero esta señal hace difícil distinguir las áreas hipointensas en varias enfermedades: cuando se producen depósitos de Calcio, hemorragias, presencia de otros metales o zonas con aire (pulmones, por ejemplo).
Lo que es más actual es el desarrollo de agentes de contraste duales, que produzcan tanto contraste T1, como contraste T2. En imagen de RM, las imágenes en T1 dan más resolución y las de T2 ayudan a detectar mejor la lesión. Por esta razón, estas sondas duales podrían dar una información muy valiosa para un diagnóstico mucho más preciso.

También se están desarrollando compuestos en los que puedan emplearse en distintas técnicas de imagen junto con la imagen de RM (PET: tomografía de emisión de positrones o SPECT: tomografía de emisión de fotón único). Serian compuestos tales como la combinación de nanopartículas de óxido de hierro con radionucleidos, como el Galio (68Ga) para PET o el Tecnecio (99Tc) para SPECT.

Imagen multimodal RM-SPECT. Al animal se le administró un compuesto dual marcado con Tecnecio y con partículas de óxido de Hierro. Para la imagen de RM se tomó la imagen en basal y después se fueron adquiriendo imágenes cada x minutos para ver el aumento de la señal tanto en los vasos como en el corazón. Para adquirir la imagen SPECT es necesario esperar un tiempo denominado captación. Las imágenes se obtuvieron a los 40 y 200 min. después de la inyección del compuesto.
Conclusiones
Terminamos la newsletter concluyendo que a día de hoy, el foco de investigación se centra en sintetizar compuestos biocompatibles de óxido de hierro, principalmente, que no produzcan toxicidad y que sean capaces de potenciar las propiedades o mejor dicho de equiparar o mejorar la resolución de las imágenes obtenidas con agentes de contraste T1, como el Gadolinio.
Si en un futuro no muy lejano conseguimos aplicar a la práctica clínica estos contrastes duales podremos utilizarlos tanto para conseguir contraste T1 y contraste T2 cuyo fin por supuesto será obtener un diagnóstico médico mucho más preciso con este tipo de sustancias.
AUTOR
Marina Benito Vicente
Responsable del Servicio de Resonancia Magnética de Investigación Hospital Nacional de Parapléjicos.
Toledo, España.
Referencia
Fernández-Barahona, I.; Muñoz-Hernando, M.; Ruiz-Cabello, J.; Herranz, F.; Pellico, J. Iron Oxide Nanoparticles: An Alternative for Positive Contrast in Magnetic Resonance Imaging. Inorganics 2020, 8, 28.
https://doi.org/10.3390/inorganics8040028












{kind=link}