En mi última newsletter, de junio de 2023, os hablé de una serie de parámetros de medición que se habían propuesto para validar modelos de inteligencia artificial para el análisis de imágenes. Quizá recordéis que os indiqué que, tanto para el entrenamiento de las redes neuronales artificiales (ANNs) como para la medición de esos parámetros, empleé los Colab Notebooks de ZeroCostDL4Mic (creados en el Henriques Lab) y prometí os explicaría un poco más de ese interesantísimo proyecto. Bueno, pues ha llegado el momento de cumplir esa promesa. Hoy os voy a explicar cómo usar ZeroCostDL4Mic de modo que podáis entrenar vuestros propios modelos de ANNs, validar los resultados y procesar imágenes con ellos. Y todo esto sin necesidad de saber programar ni tener un gran equipo para procesado de imágenes. Es suficiente con un ordenador con conexión a internet y una cuenta de Gmail.
¿Qué es ZeroCostDL4Mic?
En su página web lo definen como «un conjunto de recursos para el entrenamiento y la implementación de métodos comunes de deep learning para imágenes de microscopía. Aprovecha la facilidad de uso y acceso a la GPU proporcionada por Google Colab».
Esto quiere decir que no es necesario tener un ordenador potente con acceso a una GPU, ya que tanto el entrenamiento de la red neuronal como el análisis de las imágenes con los modelos entrenados se realiza en la nube con recursos proporcionados gratuitamente por Google. Además, tampoco es necesario saber programar en Python, ya que los Colab Notebooks de ZeroCostDL4Mic están diseñados para que solo haya que seguir las instrucciones que te van dando. Vamos a ver un ejemplo:
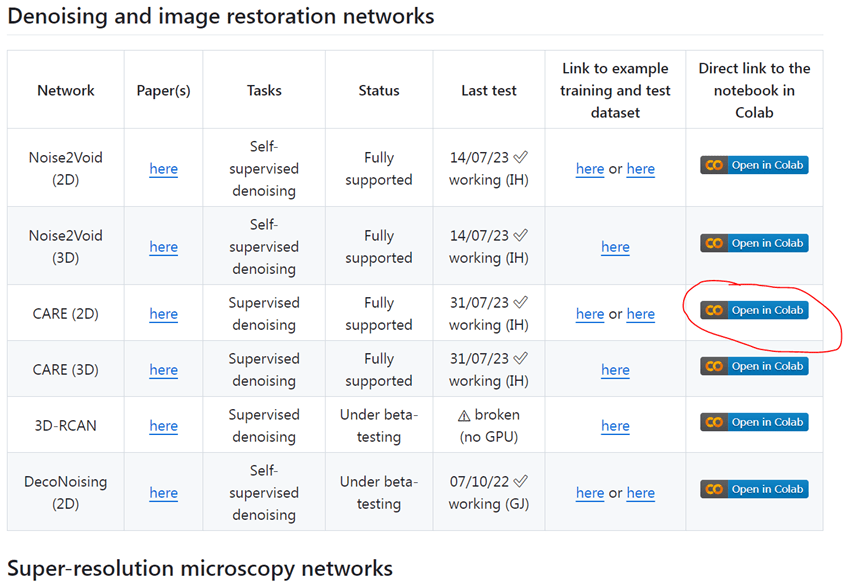
En primer lugar, abrimos el notebook que queramos usar de su página web. Para este ejemplo usaremos CARE (2D):
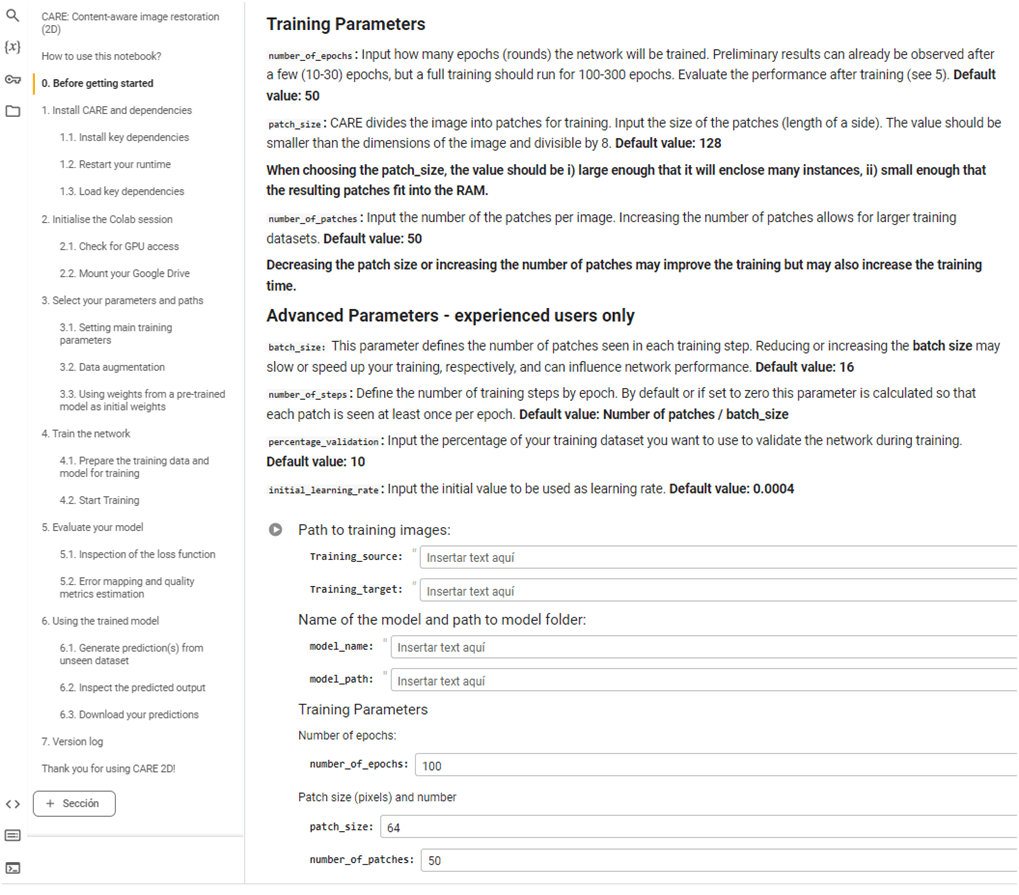
Y después, básicamente lo único que hay que hacer es seguir las instrucciones que aparecen en pantalla. La primera parte del cuaderno contiene información sobre el tipo de red neuronal que se va a usar para entrenar tu modelo y cómo citarla, instrucciones generales de cómo usar el cuaderno e instrucciones más específicas sobre cómo tienen que estar guardados los archivos de entrenamiento y validación en vuestro Drive de Google. En la columna de la izquierda se puede ver la estructura general del cuaderno.
Las siguientes celdas se tienen que ir ejecutando secuencialmente (siempre después de haber hecho lo que las instrucciones para esa celda indican). En algunas la ejecución será muy rápida, mientras que otras consumirán más tiempo.
Ejemplo de una celda en la que únicamente hay que pulsar el botón de ejecución (rodeado en rojo)
Antes de ejecutar esta celda, hay que modificar una serie de casillas, tal y como se indica en las instrucciones
Los cuadernos suelen incluir también una opción de aumento de las imágenes de entrenamiento que se puede usar en ocasiones en que se cuenta con pocas imágenes de partida. Aplica giros, volteos y otros tipos de distorsión de las imágenes originales y sus etiquetas.
Una vez entrenado el modelo (un proceso que puede llevar de minutos a horas), se puede evaluar la calidad del mismo en el propio cuaderno. Obtendremos tanto una gráfica de la loss function como distintas métricas que nos permitirán determinar el rendimiento del modelo (de las que os hablé en mi anterior newsletter).
La gráfica de la loss function nos muestra cómo a partir de las 10-20 épocas de entrenamiento se produce overfitting. Esto indica que es necesario añadir más imágenes de entrenamiento para evitar este problema y conseguir un modelo válido.
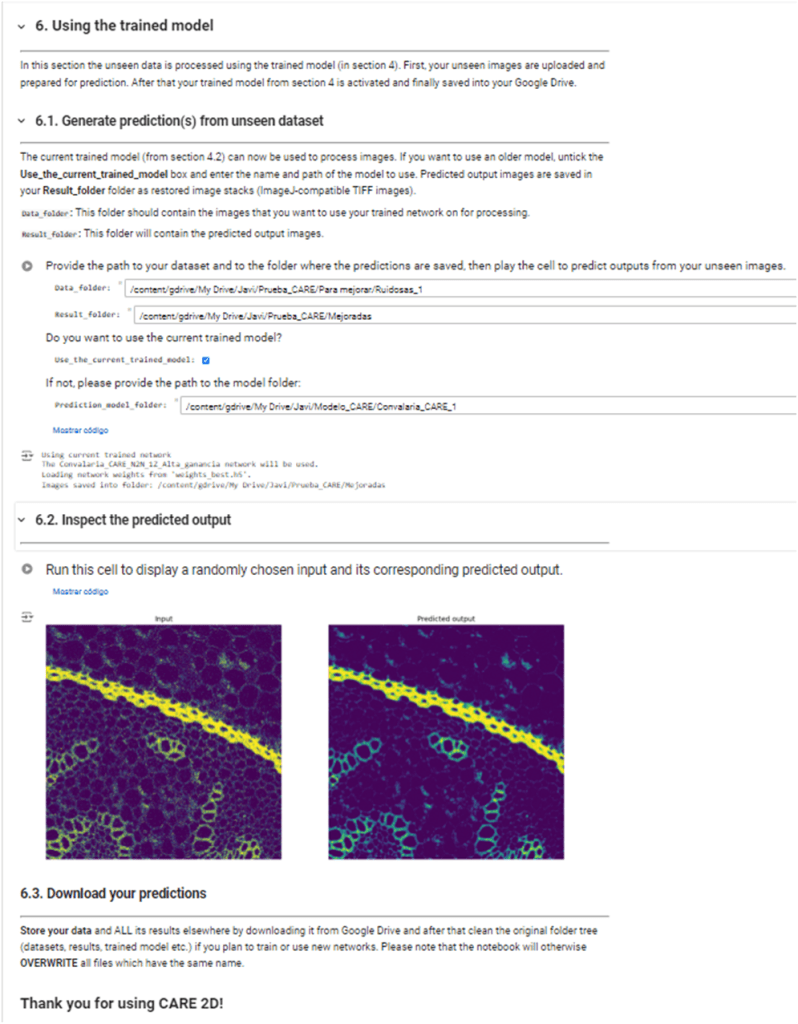
Cuando estemos contentos con los resultados obtenidos, podemos usar el modelo entrenado para procesar nuevas imágenes. Los resultados se guardarán automáticamente en nuestro Google Drive. Este paso se puede hacer también partiendo de un modelo entrenado previamente, sin necesidad de ejecutar las celdas de entrenamiento cada vez que se use el cuaderno.
Y lo único que quedará ya por hacer es descargar el modelo entrenado y las imágenes procesadas.
Así que ya veis, si sabéis leer y seguir instrucciones (en inglés), con el uso de los Colab Notebooks de ZeroCostDL4Mic tenéis el uso de la inteligencia artificial para vuestras imágenes de microscopía al alcance de la mano.
Os recomiendo que visitéis la página web del proyecto, donde podréis encontrar una serie de vídeos de demostración así como todos los Colab Notebooks disponibles y una guía paso a paso del uso de estos (más detallada que la que os he mostrado yo).
AUTOR
Javier Mazarío Torrijos Servicio de Microscopía y Análisis de Imagen Hospital Nacional de Parapléjicos. Toledo, España.
Para más información puedes contactar conmigo en: jmazario@sescam.jccm.es
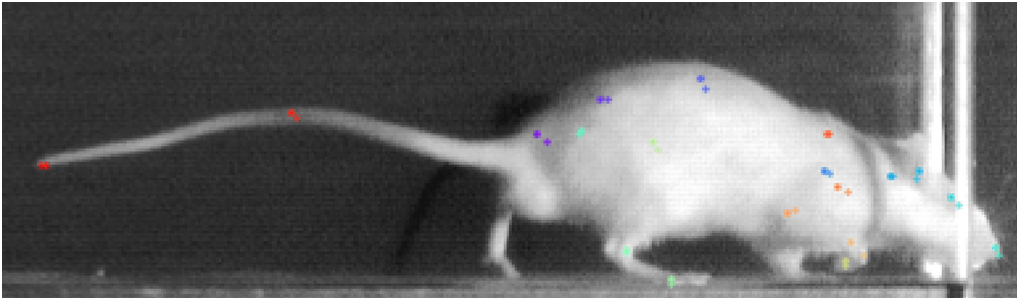
La observación del comportamiento de los animales durante los experimentos es fundamental en muchas actividades científicas, como la biomecánica, genética, etología y neurociencia. Capturar las posiciones espaciales de cada uno de los miembros del animal durante el desarrollo de una actividad sin utilizar marcadores es todo un reto. Recientemente se ha presentado una herramienta informática de última generación de código abierto llamada DeepLabCutTM(DLC) basada en algoritmos de estimación de la postura, que permite al usuario entrenar una red neuronal profunda con datos etiquetados por el usuario, para rastrear con precisión las puntos de la imagen capturada previamente definidos.
La herramienta está desarrollada como un paquete de Pyhton que incluye ciertas características como la interfaz gráfica de usuario (en inglés GraphicalUser Interface, GUI) y mejoras de rendimiento y refinamiento de la red basadas en aprendizaje activo.
En esta newsletter proporcionamos una reducida guía paso a paso que permite crear un proyecto básico en DLC usando la GUI, que nos introduzca de forma fácil y amigable en el manejo de esta interesante herramienta. Además, a día de hoy, no se ha encontrado una guía de uso de esta GUI.
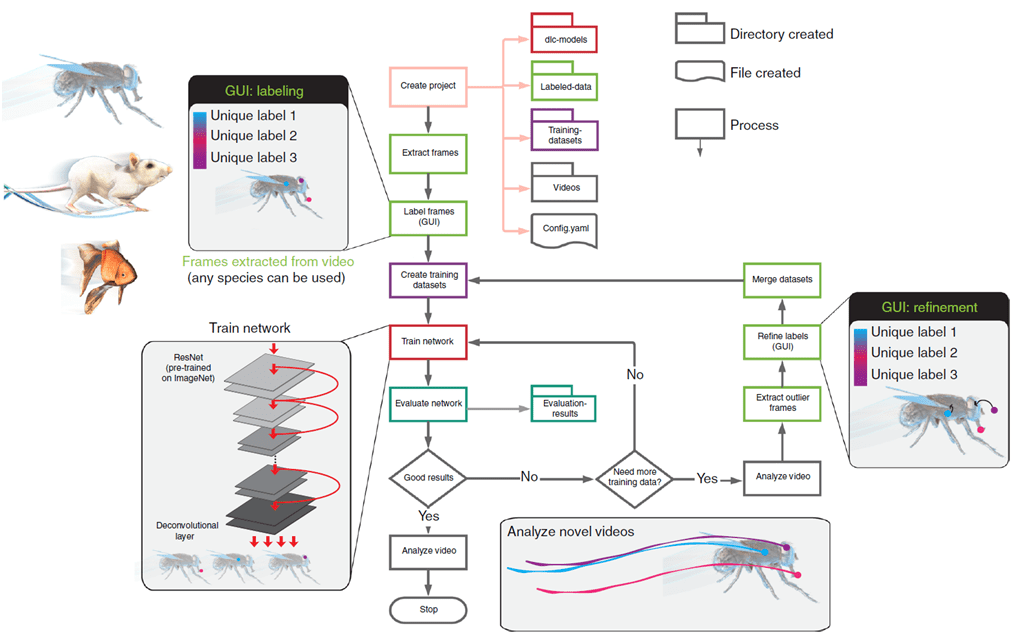
A continuación se desarrolla el flujo de trabajo de DLC (Fig. 1)sobre la GUI de la aplicación.
Fig. 2 Ventana de bienvenida de la GUI de DeepLabCut
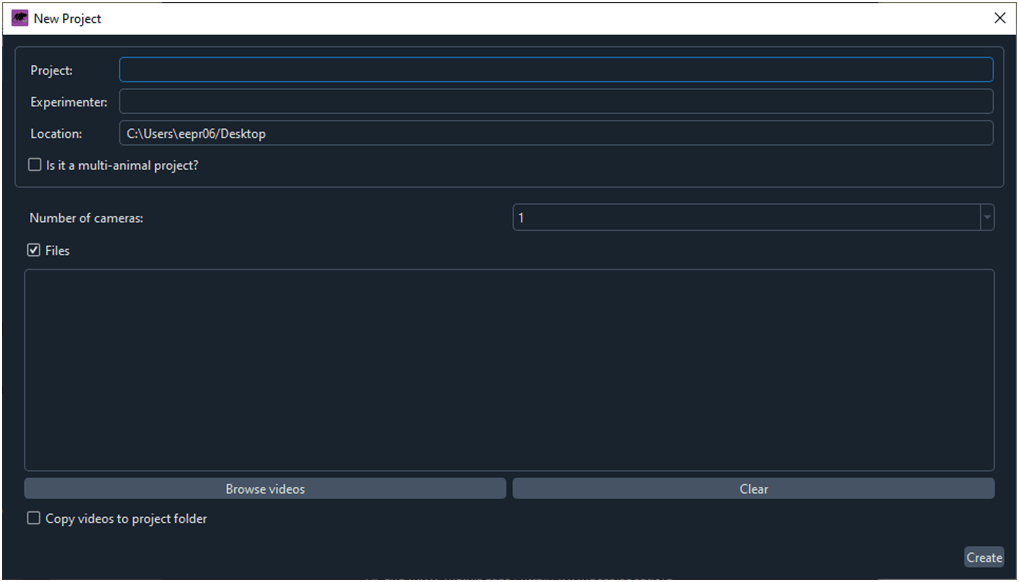
Al hacer clic sobre el botón ‘Create New Proyect’ se abre la ventana ‘New Project’ (Fig. 3).
Fig. 3 Ventana para crear un proyecto nuevo.
Introducimos el nombre del proyecto, del experimentador y de la ruta donde DLC creará una carpeta con el nuevo proyecto. Con el comando ‘Browse videos’ especificamos la carpeta donde se ubican los videos que vamos a utilizar para entrenar la red neuronal.
Al hacer clic sobre ‘Create’, DLC crea la carpeta con un nombre para el proyectoque contiene las siguientes subcarpetas y archivos:
Carpeta ‘dlc-models’: donde DLC guardará los pesos calculados para el modelo. Estos pesos son los que posteriormente DLC utilizará para predecir las variables de salida (en este caso pixel-x y pixel-y).
Carpeta ‘Labeled-data’: esta carpeta se utilizará para gestionar los fotogramas utilizados para entrenar la red.
Carpeta ‘Training-datasets’: en esta carpeta DLC guarda los datos que definen cómo se realizará el entrenamiento de la red neuronal.
Carpeta ‘Videos’: aquí se guardan los videos de donde se extraen los fotogramas para el entrenamiento.
Archivo ‘Configyaml’: es un archivo de texto editable donde el usuario especifica a DLC instrucciones acerca de la generación y aplicación del modelo.
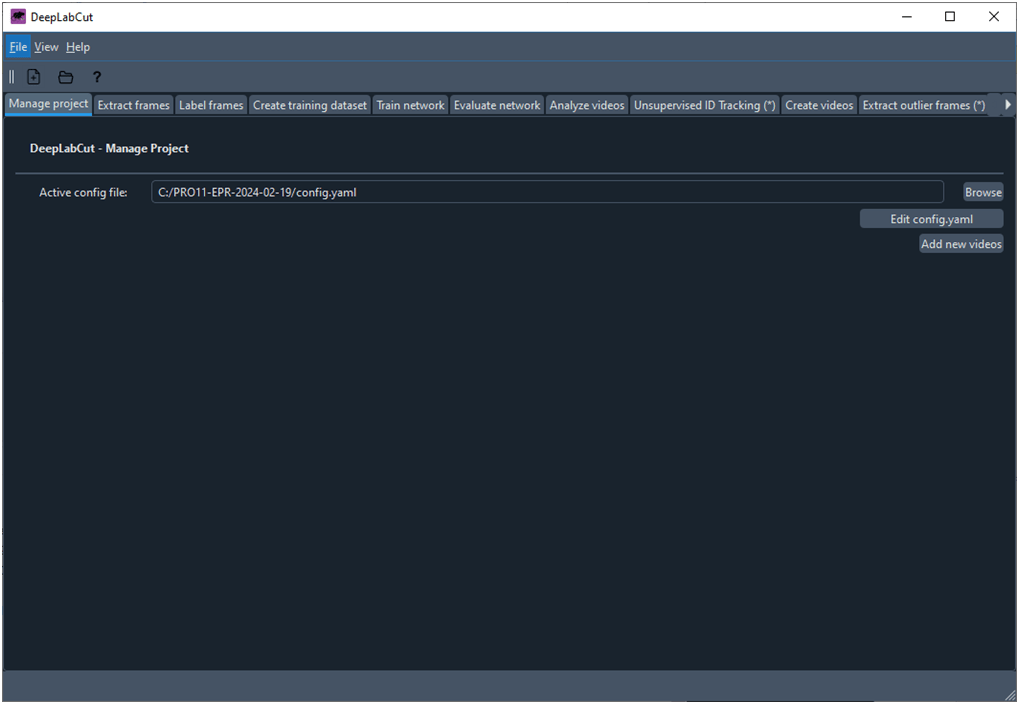
A continuación se abre la ventana principal de trabajo de DLC que contiene una serie de pestañas que dan acceso a las diferentes fichas donde el usuario controla las acciones que se realizan con DLC (Fig. 4). En concreto, una vez creado el proyecto se abre la ventana que muestra la ficha ‘Manageproject’.
Fig. 4 Ventana principal de trabajo de DeepLabCut que muestra la ficha ‘Manage Project’
A continuación si hacemos clic sobre el botón ‘Editconfig.yaml’ tendremos acceso a cambiar ciertos parámetros para desarrollar nuestro modelo:
‘bodyparts’: introducir los nombre de las etiquetas de los puntos que queremos detectar. Un ejemplo podría ser:
IliacCrest
Hip
Knee
Ankle
5 Metatarsian
‘Numframes2pikc’: introducir el número de fotogramas de cada video que vamos a etiquetar manualmente para entrenar la red.Los desarrolladores de DLC recomiendan entre utilizar entre 50 y 200 fotogramas, pero para hacer una prueba de funcionamiento podríamos reducir bastante eta magnitud.
‘TrainingFraction’: en general, un estándar admitido por la comunidad es utilizar el 70 % de datos para entrenar la red y el 30 % para validar el modelo, en este caso el valor a introducir sería 0.7. Si vamos a hacer una prueba con un reducido número de fotogramas sería mejor aumentar este parámetro, 0.95 es lo que DLC propone por defecto; esto lo deberíamos tener en cuenta a la hora de evaluar el modelo.
Obtener Fotogramas de Entrenamiento
La ficha ‘Extracframes’ contiene las instrucciones para obtener los fotogramas que DLC necesita para entrenar la red y para validar el modelo (Fig. 5). Como veremos, previamente estos fotogramas deben ser etiquetados.
Fig. 5 Ficha para obtener los fotogramas de entrenamiento.
DLC permite seleccionar entre diferentes métodos para la obtención de los fotogramas. Se pueden obtener de manera manual o automática. Si el movimiento que estamos estudiando varía a lo largo del video sin ninguna dependencia temporal el algoritmo ‘uniform’ sería la opción adecuada, si el movimiento se concentra en una parte del video, por ejemplo el estudio de un agarre, la opción ‘kmeans’ sería más adecuada.
Para una prueba estándar se pueden dejar los parámetros de la Fig. 5, que DLC propone por defecto.Con la opción ‘kmeans’ nos aseguramos de que los fotogramas no van a ser parecidos, lo cual es muy conveniente para el entrenamiento.
Al hacer clic sobre ‘ExtractFrames’,DLC crea una subcarpeta por cada video de entrenamiento en la carpeta ‘Labeled-data’. Cada subcarpeta contendrá los fotogramas del video seleccionados para el entrenamiento.
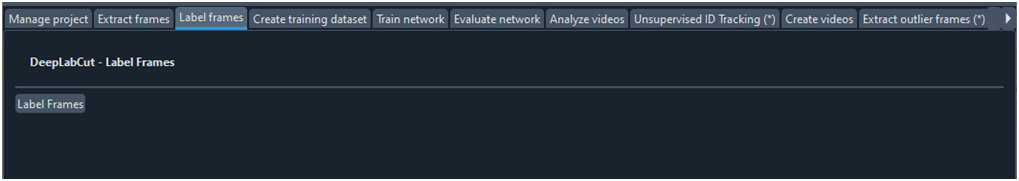
Etiquetar fotogramas
Haciendo clic sobre ‘Labelframes’ (Fig. 6) DLC nos pregunta por la subcarpeta ubicada en ‘Labeled-data’ donde se encuentran los fotogramas que deseamos etiquetar. Realizado la anterior acción se abre la GUI de etiquetado de fotogramas (Fig. 7).
Fig. 6 Ficha que da acceso a la GUI de etiquetado
Fig. 7 GUI de etiquetado de fotogramas
En la parte inferior derechade la GUI de etiquetado aparecen las etiquetas (‘keypoints’ o ‘bodyparts’) que introdujimos en el archivo ‘config.yaml’. Utilizar los controles en la parte superior izquierda para etiquetar: la diana para ubicar una etiqueta y la ‘x’ para eliminar una etiqueta. Con la barra inferior nos movemos entre los diferentes fotogramas. Finalmente, antes de cerrar la ventana es necesario salvar los datos.
Crear conjunto de datos de entrenamiento
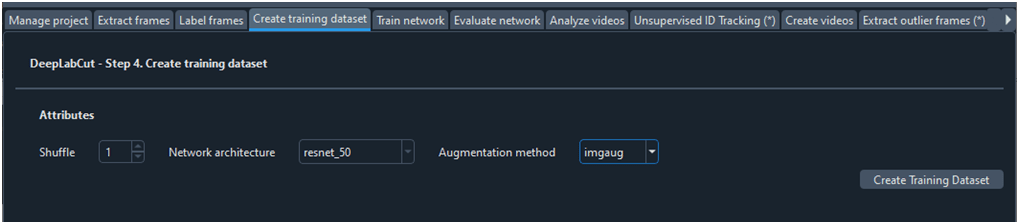
Para entrenar la red neuronal es necesario crear un conjunto de datos de entrenamiento. De todos los fotogramas etiquetados DLC toma un grupo para entrenar la red y otro grupo para validar el modelo, según ‘TrainingFraction’ en ‘config.yaml’. Podemos dejar la ficha ‘Create training dataset’ con la configuración por defecto (Fig. 8). El campo ‘Shuffle’ es para diferenciar entre modelos y poder compararlos. Con el parámetro ‘imgaug’ se expande artificialmente el conjunto de entrenamiento aplicando varias transformaciones a las imágenes (por ejemplo, rotación o cambio de escala) para generar modelos más robustos y precisos.
Fig. 8 Ficha para crear un conjunto de datos de entrenamiento
Entrenar la red neuronal
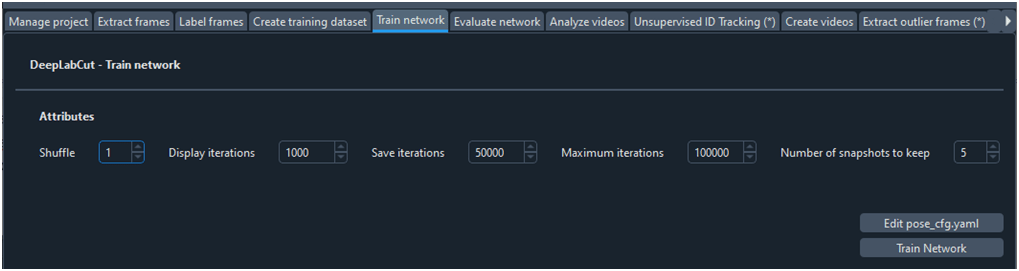
Para entrenar la red neuronal utilizamos la ficha ‘Train network’ (Fig. 9). Al hace clic sobre el botón ‘Train Network’ comienza un proceso de cálculo paramétrico iterativo y continuo basado en árboles de decisión durante el cual se van actualizando los pesos de los parámetros con los que luego se realizarán la predicciones. Se puede visualizar la pérdida del modelo cada ciertas iteraciones (‘Displayiterations’), se puede guardar el modelo cada ciertas iteraciones (‘Saveiterations’) y se puede configurar el número máximo de modelos que queremos guardar (‘number of senashots to keep’). También se debe configurar el máximo número de iteraciones que queremos realizar para entrenar la red (‘Maximumiterations’).
Fig. 9 Ficha para entrenar una red neuronal
El proceso de entrenar la red puede demorarse varias horas, multiplicándose por 10 en caso de que no dispongamos de una tarjeta gráfica dedicada.
Evaluar la red neuronal
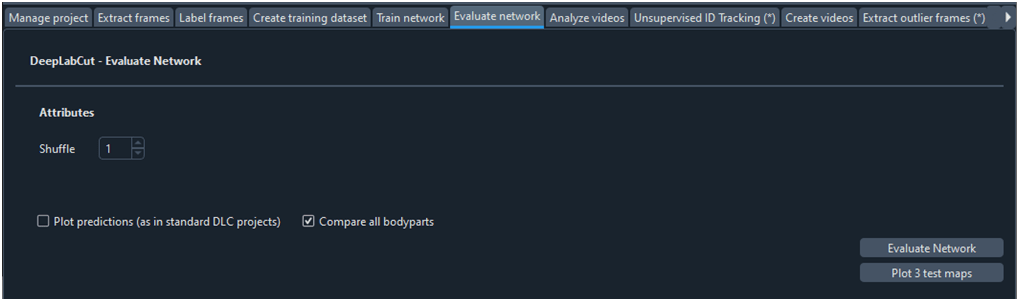
Es importante evaluar la bondad de la red entrenada. En este paso DLC aplica el modelo a todos los fotogramas, tanto a los de entrenamiento como a los de validación, y calcula el error y la probabilidad para ambos grupos.Al hacer clic sobre el botón ‘Evaluate Network’ de la ficha ‘Evaluatenetworw’ (Fig. 10) DLC crea la carpeta ‘evaluation-results’ dentro de la carpeta del proyecto. Esta carpeta contiene un archivo ‘.csv’ con los resultados de la evaluación (Fig. 11), si se selecciona la opción ‘Plotpredictions’ se genera una carpeta con los fotogramas de entrenamiento y de test en los que se muestran las etiquetas de entrenamiento y las estimadas por el modelo (Fig. 12).
Fig. 10 Ficha para evaluar la red neuronal
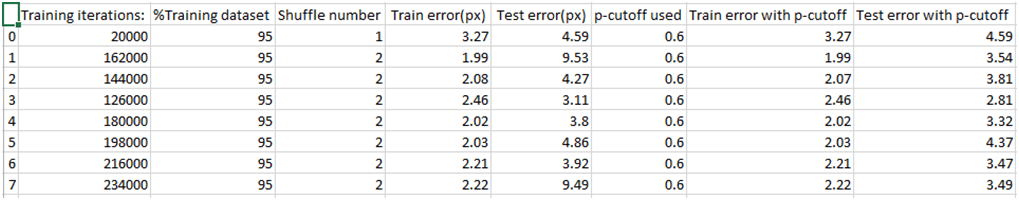
Fig. 11 Hoja de resultados de la evaluación de diferentes modelos
Fig. 12 Ejemplo de fotograma mostrando las etiquetas de entrenamientos y las estimadas por el modelo
Analizar videos
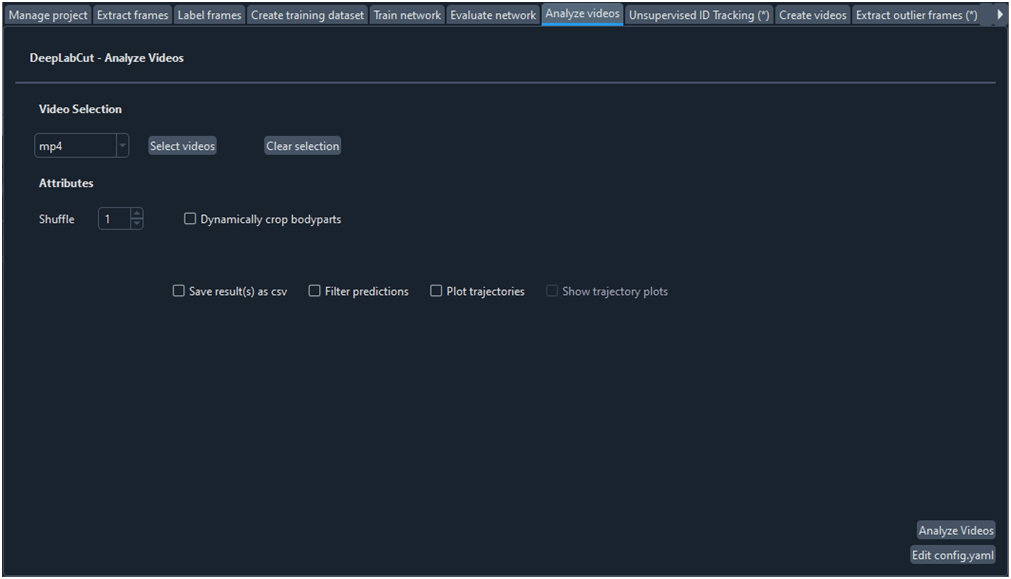
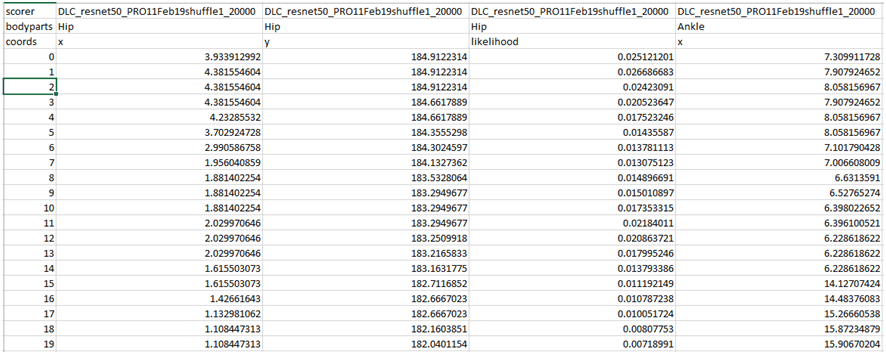
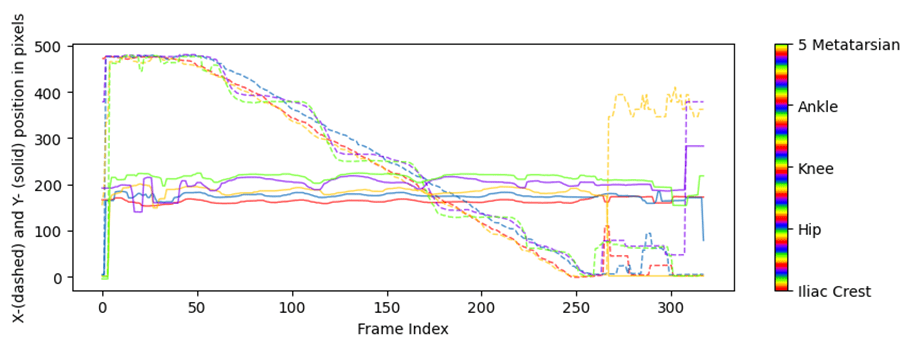
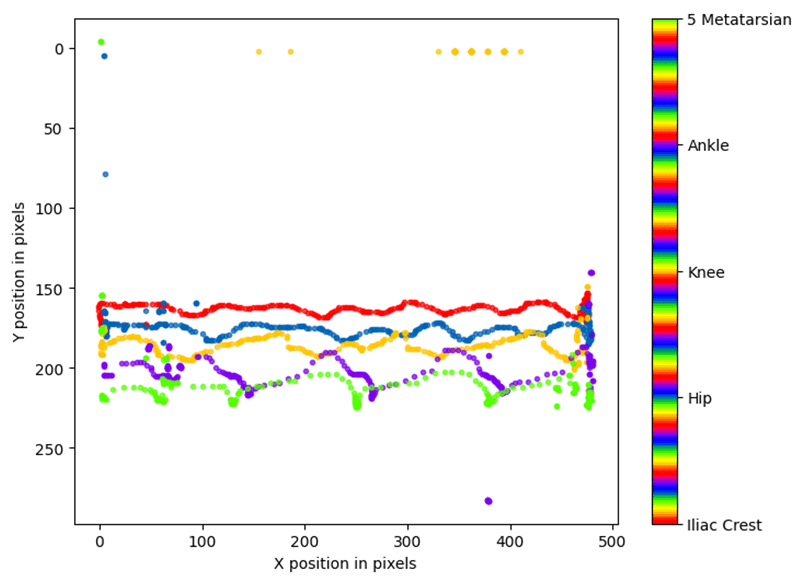
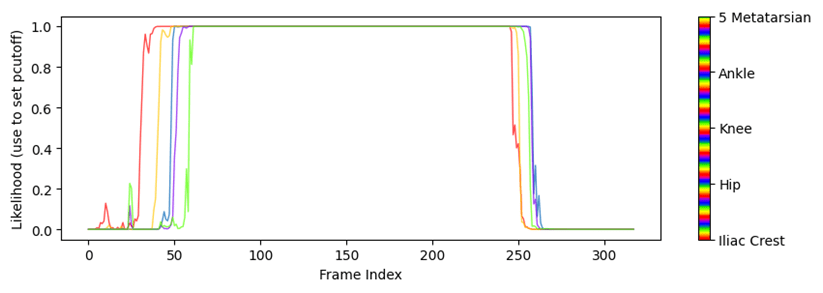
Para obtener las coordenadas xyde un vídeo o conjunto de vídeos hacemos uso de la ficha ‘Analyze videos’ (Fig. 13). Con esta función DCL aplica el modelo a cada fotograma del video en cuestión, calcula las coordenadas x e y en píxeles y la probabilidad de que el pixel estimado se encuentre donde se ha calculado. Al hacer clic sobre el botón ‘Analize Videos’ DLC crea en la carpeta donde se encuentra el video a analizar, además de otros archivos, un archivo ‘csv’ con los valores anteriormente comentados para cada fotograma y para cada etiqueta (o ‘bodyparts’ en config.yaml) (Fig. 14). Marcar la opción ‘Filterpredictions’ para obtener un archivo ‘csv’con los datos filtrados. La opción ‘Plottrajectories’ proporciona cuatro gráficos muy útiles para visualizar el análisis del video (Fig. 15, Fig. 16, Fig. 17 y Fig. 18).
Fig. 13 Ficha para analizar un vídeo
Fig. 14 Archivo ‘csv’ con las coordenadas x-pixel e y-pixel y la probabilidad para cada fotograma y para cada etiqueta
Fig. 15 Coordenadas ‘x-pixel’ e ‘y-pixel’ frente al tiempo
Fig. 16 Posiciones de las ‘bodyparts’
Fig. 17 Histograma de diferencias entre posiciones consecutivas de ‘bodyparts
Fig. 18 Probabilidad de las ‘bodyparts’ frente al tiempo
Referencias
Nath, T., Mathis, A., Chen, A.C. et al. Using DeepLabCut for 3D markerless pose estimation across species and behaviors. Nat Protoc 14, 2152–2176 (2019).
La Unidad de Investigación del Hospital Nacional de Parapléjicos está adherida al acuerdo COSCE de transparencia sobre el uso de animales de experimentación científica en España desde su creación en el año 2.016. Fuimos el primer Hospital en adherirse y durante mucho tiempo los únicos, como única referencia en el apartado de ciencia en sistemas de salud.
Siempre entendimos desde el Servicio de Animalario y Cirugía Experimental la importancia de la transparencia en la investigación con animales.
El Acuerdo es un compromiso de actuación de cuatro puntos:
PRIMER COMPROMISO
Hablar con claridad sobre cuándo, cómo y por qué se usan animales en investigación
SEGUNDO COMPROMISO
Proporcionar información adecuada a los medios de comunicación y al público en general sobre las condiciones en las que se realiza la investigación que requiere el uso de modelos animales y los resultados que de ella se obtienen
TERCER COMPROMISO
Promover iniciativas que generen un mayor conocimiento y comprensión en la sociedad sobre el uso de animales en investigación científica
CUARTO COMPROMISO
Informar anualmente sobre el progreso y compartir experiencia
La Confederación de Sociedades Científicas de España (COSCE) propuso en 2016 a la comunidad científica española el Acuerdo COSCE de transparencia sobre el uso de animales de experimentación científica en España con el objetivo de mejorar el nivel de entendimiento por parte de la sociedad sobre los beneficios, perjuicios y limitaciones que la experimentación con animales puede conllevar.
El Acuerdo se presentó a las instituciones españolas relacionadas con la experimentación animal, incluyendo tanto a las que crían y/o utilizan animales en investigación como a las que agrupan a los profesionales que de algún modo tienen que ver con el cuidado y uso de los animales, y también asociaciones de pacientes. La propuesta de COSCE se concretaba en una invitación a todas las organizaciones a firmar el Acuerdo y por lo tanto a cumplir de forma voluntaria sus cuatro compromisos.
Los tres primeros compromisos del Acuerdo COSCE se centran en las actividades de comunicación interna y externa relacionadas con la utilización de animales, y el cuarto compromiso en la información del progreso del Acuerdo y la puesta en común de las experiencias desarrolladas.
El Acuerdo tuvo una acogida muy positiva y fue presentado en septiembre de 2016 ya con la firma de numerosas instituciones. La adhesión de nuevas instituciones continúa, y en la fecha de publicación de este informe hay 166 instituciones adheridas. El Acuerdo de Transparencia en España es el que tiene más instituciones adheridas de todo el mundo, y está sirviendo de ejemplo para al desarrollo de acuerdos de este tipo en otros países.
La encuesta anual, que recoge las acciones de apertura y transparencia de las instituciones españolas en 2022, se han llevado a cabo, como en los cinco años anteriores, de forma independiente por la European Animal ResearchAssociation (EARA).
Las encuestas fueron completadas por 159 instituciones, 104 que utilizan animales y otras 55 que no. Cinco instituciones no han contestado. Otras cinco instituciones de nueva incorporación en 2023, han contestado sin tener la obligación de hacerlo todavía.
Esta última encuesta describe y analiza las respuestas obtenidas en la encuesta de forma ordenada para cada uno de los compromisos del Acuerdo COSCE, que confirman la continuación de la tendencia observada en años anteriores hacia un incremento de las acciones transparencia.
La evidencia más clara de la consolidación de actividades de transparencia es el mantenimiento por todas las organizaciones adheridas al Acuerdo COSCE de la declaración institucional en su página web donde se explica su política y condiciones respecto al uso de animales en investigación. También se han recogido otros ejemplos de actividades de transparencia promovidos por el Acuerdo, como visitas de estudiantes o periodistas a algunos establecimientos usuarios de animales, o numerosas participaciones proactivas en medios de comunicación.
Algunas informaciones destacadas de las encuestas han sido:
● 159 instituciones de diferentes tipologías y de todo el ámbito nacional han completado la encuesta, lo que supone el informe anual con la mayor participación de instituciones en el mundo. Representa un incremento de 12 centros respecto al año anterior.
● El 100% dispone en su web de una declaración institucional sobre el uso de animales de experimentación, y el 82% tiene esta declaración visible en tres o menos clics desde la página principal.
● El 72% ha publicado noticias en la web de su institución relacionadas con la investigación con animales
● El 76% ha participado en actividades de divulgación científica (un 12% más que en el año anterior) y el 86% ha proporcionado acceso a visitantes externos (un 5% más respecto al año anterior).
● Desde la toma de los primeros datos en 2018, ha aumentado la referencia al uso de animales en investigación en relación con los medios de comunicación del 37% al 67%.
● El 17% tiene una política institucional sobre la comunicación de los modelos animales que se utilizan en investigación en notas de prensa y comunicados, y un 34% tiene intención de implementarla en el futuro.
● Solamente 6 instituciones han manifestado dificultades en el cumplimiento del segundo compromiso en 2022, debidas a la falta de recursos para realizar formaciones específicas sobre los medios de comunicación y falta de tiempo para llevar a cabo iniciativas en este aspecto.
El Acuerdo COSCE de transparencia sobre el uso de animales en experimentación científica en España ha tenido repercusión internacional, y varios representantes del mismo han sido invitados a presentarlo en distintos foros y congresos internacionales durante 2022, como por ejemplo el Congreso de la Federation of EuropeanLaboratory Animal ScienceAssociations (FELASA), Expobioterios en Latinoamérica, la reunión anual de miembros de AAALAC International, y la Jornada Científica de la Sociedad Española para las Ciencias del Animal de Laboratorio (SECAL). Además, un miembro de la Comisión COSCE de animales en experimentación científica ha colaborado en las reuniones para el desarrollo de un Acuerdo de este tipo (USARO) en los Estados Unidos. En la actualidad hay Acuerdos de Transparencia activos en ocho países de Europa (Reino Unido – 2014 // España – 2016 // Portugal – 2018 // Bélgica – 2019 // Francia, Alemania y Países Bajos – 2021 // Suiza – 2022), también en Nueva Zelanda – 2021 y recientemente en Australia – 2023. Y se está trabajando para su implantación en muchos otros incluyendo los Estados Unidos.
Respecto a la valoración general que hacen las instituciones sobre el sexto año del Acuerdo de Transparencia:
Prácticamente la totalidad (98,7%) considera que es un paso importante para la investigación biomédica en España.
El 99,3% de las instituciones declaran entender los cuatro compromisos para su cumplimiento.
Un 90% declara tener el apoyo suficiente para implementar el Acuerdo por parte de su institución (el año anterior fue el 85%) y un 78% cree que en las organizaciones firmantes puede haber cambios reales (20% en el año anterior).
CONCLUSIÓN
La información por parte del personal implicado en la investigación con animales y otras materias sensibles, que conocen de primera mano todos los aspectos relacionados con la misma, es la forma de que la opinión pública tenga un conocimiento claro y real de lo que hacemos en investigación. Se evita sí la distorsión que distintos medios y asociaciones antiviviseccionistas puedan llevar a cabo en la opinión pública, muchas veces derivadas de su profundo desconocimiento sobre la realidad de la investigación.
Nuestro país ha sido pionero en esta iniciativa, que han adoptado otros países europeos, con continuas adhesiones tanto de nuevas instituciones como de nuevos estados.
AUTOR
Enrique Páramo Rosel Responsable del Servicio de Animalario y Cirugía Experimental Hospital Nacional de Parapléjicos. Toledo, España.
En esta newsletter queremos hablaros un poco sobre una técnica analítica ampliamente utilizada, la espectrometría de masas por imágenes (IMS).
Comprender la biodistribución, el metabolismo y la acumulación de fármacos en el cuerpo es un elemento fundamental en investigacióny enel desarrollo farmacéutico.LaIMSes una tecnología de vanguardia que puede proporcionar una amplia información molecular/química sobre biomoléculas, complementaria a la información que se obtiene con otros métodos de análisis espacial de muestras biológicas (Hematoxilina/ eosina (H&E), inmunohistoquímica (IHC)). Su objetivo es detectar los componentes moleculares (metabolitos, péptidos, proteínas) de la muestra, que pueden correlacionarse con las características histológicas y, en última instancia, proporcionar un vínculo molecular fenotípico con la compleja biología de una patología.
Introducción.
Desde el final de los años 90, cuando se empezó a utilizar la espectrometría de masas MALDIpara analizar secciones de tejidos de mamíferos y producir imágenes iónicas de péptidos y proteínas,se ha confirmado la habilidad de cartografiar moléculas particulares en coordenadas bidimensionales, dando lugar a la creación de imágenes moleculares.
Entre las diversas técnicas de ionización, MALDI IMS es líder en el análisis de distribuciones moleculares en diversos tejidos y diferentes enfermedades. MALDI IMS es capaz de cartografiar biomoléculas de interés con alta resolución espacial (∼10 μm) y alta sensibilidad.
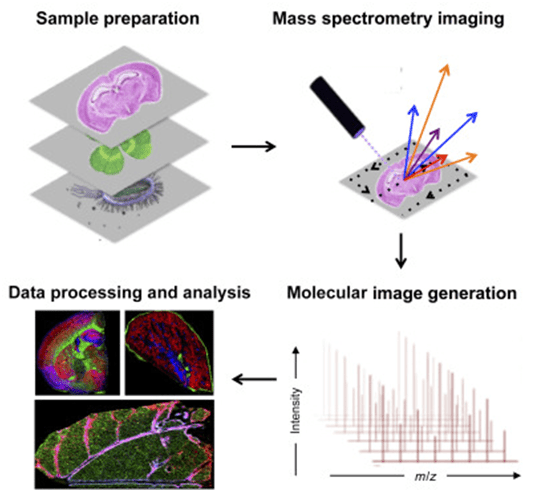
Flujo de trabajo para la adquisición de imágenes por espectrometría de masas.
¿Qué tipo de muestras puedo utilizar?
Pueden utilizarse muestras de tejido fresco congelado, pero también muestras de tejido fijadas en formalina e incluidas en parafina (FFPE), incluidos los microarrays de tejidos (TMA).
Figura 1 Visión general del flujo de trabajo en la obtención de imágenes por espectrometría de masas.[8]
La preparación y conservación de las muestras son los primeros pasos críticos para obtener imágenes iónicas de excelente calidad durante un experimento MALDI IMS. La estandarización de los procedimientos de preparación es necesaria para obtener resultados reproducibles, especialmente cuando se manejan grandes conjuntos de muestras o en un entorno clínico.
Respecto a la preparación de muestra, no existe un método que se adapte a todos los tejidos y analitos. Cada paso del proceso dentro del flujo de trabajo debe evaluarse y optimizarse en función de los tejidos, las hipótesis y los objetivos de investigación.
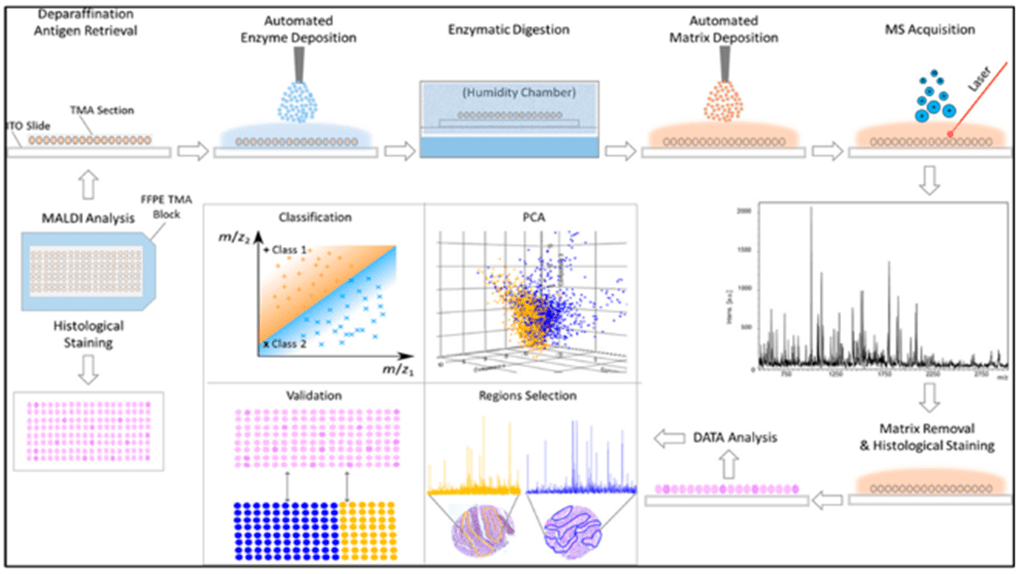
Figura 2 Flujo de trabajo para muestras FFPE TMA para detección de proteínas
El flujo de trabajo de MALDI IMS incluye una serie de pasos distintos que comienzan con el seccionamiento del tejido, que puede variar entre 5 y 15 μm de grosor dependiendo del tejido. El lavado del tejido para eliminar sales y lípidos interferentes es vital para el análisis de proteínas. La elección de matrizse realizará en función del tipo de analito objeto de estudio, ya que el éxito del análisis depende de que la matriz elegida favorezca una ionización y fragmentación eficiente del mismo. A continuación, el espectrómetro de masas recoge de forma automatizada espectros de masas de ubicaciones únicas a lo largo de la superficie de la muestra, y cada ubicación de muestreo contiene un espectro de masas único. Los espectros de masas adquiridos se combinan y procesan mediante algoritmos de software para crear una imagen MS analizable.
Tras la adquisición por MS, puede retirarse la matriz y teñir la muestra restante. Las secciones de tejido teñidas se utilizan para definir áreas de interés.
Aplicaciones.
MALDI IMS combina una sensibilidad y selectividad sin precedentes gracias a la espectrometría de masas de alta resolución, que puede combinarse con la histología tradicional. Esta tecnología única ha ganado popularidad en los últimos años en diversos campos de investigación, como la investigación médica, las plantas, los hongos y las bacterias.
Investigación médica:
Tejidos: MALDI IMS se ha aplicado con éxito a varios tejidos diferentes y en la investigación médica de diversas enfermedades. Uno de los campos en los que más se ha investigado es el cáncer, pero en los últimos años se postula como herramienta emergente en neurología para comprender mejor la patología celular de la enfermedad y/o su gravedad[3][4].
Aplicada a lesión medular se han detectado y localizado de forma específica acilcarnitinas de cadena larga a lo largo de los márgenes de la lesión a los 3 días de inducir la lesión y manteniéndose entre 7 y 10 días. [5].
En estudios de enfermedades relacionadas con el dolor, se ha investigado la distribución de neuropéptidos en médula (predominantemente localizados en la médula espinal dorsal) relacionados con el dolor. [6].
Otro ejemplo lo encontramos en el estudio de desmielinización de la médula espinal murina[7] en el que determinaron los cambios lipídicos que ocurren principalmente dentro de las lesiones en la desmielinización primaria.
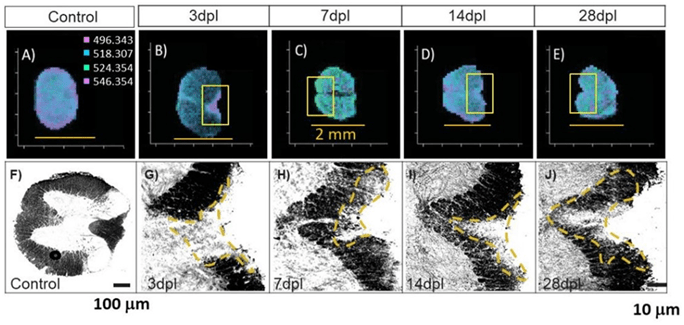
Figura 3 MALDI MSI de (A) secciones de médula espinal lesionadas, (B) 3 dpl, (C) 7 dpl, (D) 14 dpl y (E) 28 dpl (barras de escala = 2 mm) y el tejido teñido correspondiente de secciones de médula espinal (F) no lesionadas, (G) 3 dpl, (H) 7 dpl, (I) 14 dpl y (J) 28 dpl. Las inserciones en caja de los paneles B-E corresponden a las imágenes teñidas G-J.
Células:Además de los tejidos, la obtención de imágenes celulares por MALDI IMS ha ido ganado popularidad; se ha trabajado mucho en esferoides y se está trabajando mucho en la obtención de imágenes unicelulares.
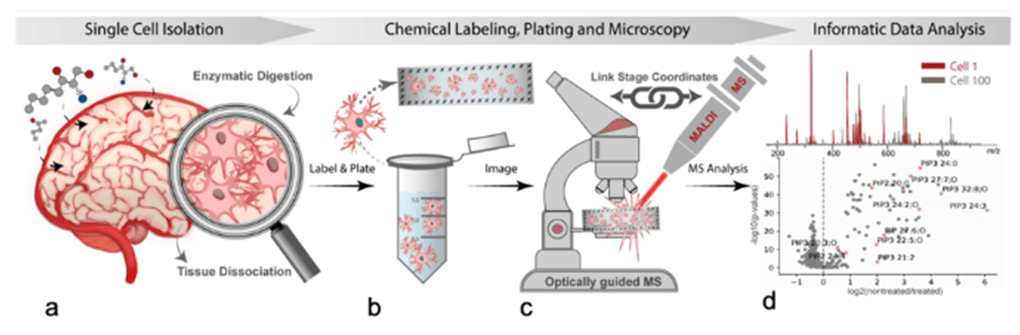
Figura 4 Flujo de trabajo de células individuales por espectrometría de masas MALDI guiada por imágenes. (a) Una región tisular aislada se somete a digestión enzimática para eliminar la matriz extracelular y los componentes conectivos, liberando las células individuales en la solución. (b) A continuación, las células se tiñen con una tinción nuclear y la solución de células individuales se extiende a través de un portaobjetos de microscopía que tiene marcadores fiduciales grabados alrededor del perímetro. (c) Se realiza una microscopía de campo claro y fluorescencia para determinar la ubicación relativa de las células en el portaobjetos. Se registran las coordenadas de píxel de los marcadores fiduciales y se inserta el portaobjetos en el espectrómetro de masas MALDI. Utilizando el punto de mira de la cámara MALDI, se encuentran los mismos puntos fiduciales y se registran sus coordenadas. A continuación se realiza el registro de la imagen utilizando los marcadores fiduciales para traducir las coordenadas de píxel de las células individuales a las coordenadas físicas de la platina del espectrómetro de masas. (d) Espectrometría de masas MALDI de alto rendimiento y análisis de estilo informático en el que cada adquisición MALDI representa una única célula. Imagen inédita, cortesía del Dr. Jonathan Sweedler y Dan Castro de la Universidad de Illinois Urbana-Champaign.
Ventajas y Desventajas
Ventajas.
Capacidad para detectar, cuantificar relativamente y mapear moléculas pequeñas (<2kDa) y los metabolitos dentro de una muestra.
Alta resolución espacial y especificidad molecular sin necesidad de marcadores químicos, procedimientos de tinción y sondas moleculares.
Complemento a las técnicas de histología.
Desventajas.
Las principales desventajas que presenta derivan del proceso de preparación de la muestra.
Supresión de iones: ocurre cuando las naturalezas químicamente distintas de cada metabolito perjudican la detección general. Los analitos más abundantes se ionizan selectivamente sobre los menos abundantes.
Degradación de la muestra, que puede ocurrir cuando el tejido se descongela durante la preparación de la muestra.Ciertos analitos no son estables a temperatura ambiente y pueden degradarse por completo, lo que lleva a una interpretación errónea de los datos.
La aplicación no uniforme de la matriz puede producir artefactos.
No existe una estandarización de la preparación de la muestra.
Los métodos alternativos de ionización ya han demostrado ser eficaces para eludir algunos de estos problemas. Por ejemplo, la MS de iones secundarios minimiza los problemas con las inconsistencias de la matriz y la difusión del analito al no requerir la aplicación de la matriz en absoluto.
Conclusiones
MALDI IMS sigue avanzando en la comprensión científica de las enfermedades al proporcionar información espacial sobre los analitos dentro de los sistemas biológicos. Comprender dónde se encuentran las biomoléculas en relación con la patología y en relación con otras biomoléculas permite a los investigadores un conocimiento profundo del sistema. En las últimas décadas, se ha puesto énfasis en el avance de esta técnica para encontrar alternativas que aumenten la sensibilidad de la detección, mejoras en la instrumentación y una mayor comprensión en la preparación de muestras. Estos avances han llevado a reforzar el uso de MSI para el análisis de terapias y biomoléculas.
Bibliografía
[1] J. L. Moore and G. Charkoftaki, “A Guide to MALDI Imaging Mass Spectrometry for Tissues,” J. Proteome Res., vol. 22, no. 11, pp. 3401–3417, 2023, doi: 10.1021/acs.jproteome.3c00167.
[2] J. H. Holbrook, G. E. Kemper, and A. B. Hummon, “Quantitative mass spectrometry imaging: therapeutics & biomolecules,” Chem. Commun., pp. 2137–2151, 2024, doi: 10.1039/d3cc05988j.
[3] K. Chen, D. Baluya, M. Tosun, F. Li, and M. Maletic-Savatic, “Imaging mass spectrometry: A new tool to assess molecular underpinnings of neurodegeneration,” Metabolites, vol. 9, no. 7. 2019. doi: 10.3390/metabo9070135.
[4] L. K. Schnackenberg, D. A. Thorn, D. Barnette, and E. E. Jones, “MALDI imaging mass spectrometry: an emerging tool in neurology,” Metab. Brain Dis., vol. 37, no. 1, pp. 105–121, 2022, doi: 10.1007/s11011-021-00797-2.
[5] J. Quanico et al., “3D MALDI mass spectrometry imaging reveals specific localization of long-chain acylcarnitines within a 10-day time window of spinal cord injury,” Sci. Rep., vol. 8, no. 1, p. 16083, Dec. 2018, doi: 10.1038/S41598-018-34518-0.
[6] P. Sui et al., “Neuropeptide imaging in rat spinal cord with MALDI-TOF MS: Method development for the application in pain-related disease studies,” http://dx.doi.org/10.1177/1469066717703272, vol. 23, no. 3, pp. 105–115, May 2017, doi: 10.1177/1469066717703272.
[7] E. R. Sekera, D. Saraswat, K. J. Zemaitis, F. J. Sim, and T. D. Wood, “MALDI Mass Spectrometry Imaging in a Primary Demyelination Model of Murine Spinal Cord,” J. Am. Soc. Mass Spectrom., vol. 31, no. 12, pp. 2462–2468, Dec. 2020, doi: 10.1021/JASMS.0C00187/SUPPL_FILE/JS0C00187_SI_001.PDF.
[8] M. de Raad, T. R. Northen, and B. P. Bowen, “Analysis and Interpretation of Mass Spectrometry Imaging Datasets,” Compr. Anal. Chem., vol. 82, pp. 369–386, Jan. 2018, doi: 10.1016/BS.COAC.2018.06.006.
AUTOR
Gemma Barroso García, MSc. Responsble SAI-Proteómica. Hospital Nacional de Parapléjicos. Toledo, España.
Para más información, dudas o solicitud de presupuestos puedes contactarnos en: unidadproteomica.hnp@sescam.jccm.es
Como ya conocéis de seminarios, jornadas y newsletter anteriores (NL21-CITF_04 CITOMETRIA DE FLUJO Y VEs EN LESIÓN MEDULAR Y ENFERMEDADES NEURODEGENERATIVAS), las Vesículas Extracelulares (EVs) son unas partículas secretadas por todas las células, que actúan como mecanismo de comunicación intercelular y que tienen un papel importante tanto en procesos fisiológicos como patológicos.
Podemos encontrar estas EVs en todos los fluidos, desde el plasma hasta incluso la leche materna, pudiendo tratarse como posibles biomarcadores en biopsia líquida. Pero lo cierto es que se liberan al medio extracelular y que, por tanto, podemos encontrarlas también dentro de un tejido, donde ejercerán diversas funciones a nivel local.
PROTOCOLO PARA EL AISLAMIENTO/ENRIQUECIMIENTO DE EVs
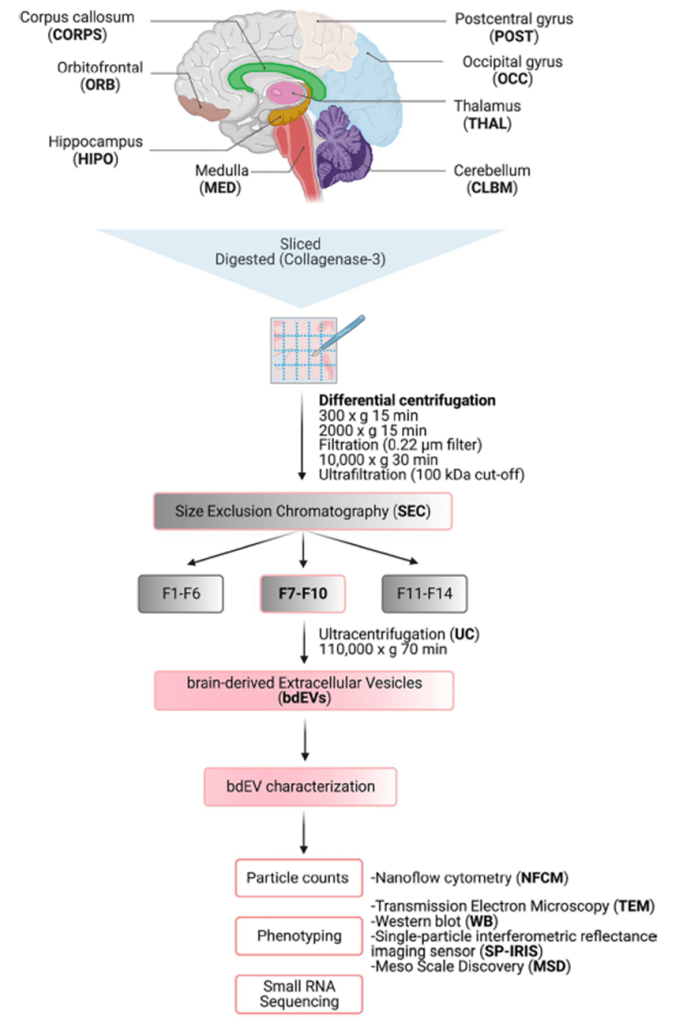
En este artículo los autores utilizan un único cerebro de un varón de 22 años, cuyas regiones fueron diseccionadas y congeladas a -80ºC con un delay de 13h postmortem. Por tanto, el aislamiento de las EVs tuvo lugar a partir de tejido congelado.En la fig.1 se recogen todos los pasos del procedimiento.
En primer lugar, y como ocurre con la mayor parte de los protocolos para obtención de células del sistema nervioso, se realizó una digestión enzimática, en este caso con colagenasa.
Es importante tener en cuenta que durante la digestión de cualquier tejido, se va a producir muerte celular por el mero procedimiento, con lo que el homogenado resultante, además de EVs contendrá cuerpos apoptóticos y otros restos celulares que en el artículo que nos ocupa, los autores intentan minimizar con los siguientes pasos del protocolo.
El siguiente paso del protocolo consistió en un par de centrifugaciones diferenciales secuenciales: la primera a 300 x g por 10 minutos, con la finalidad de retirar las células (que se pueden utilizar para otro tipo de caracterizaciones por citometría), y el sobrenadante de esta primera centrifugación se centrifuga de nuevo a 2.000 x g 15 minutos, para eliminar los cuerpos apoptóticos y restos celulares grandes.
Con el fin de eliminar lo más posible el debris celular, realizaron una filtración del sobrenadante de la última centrifugación con un filtro de 0.22 µm, y a continuación centrifugaron a 10.000 x g durante 30 min. A esta velocidad de centrifugación, el pellet que se obtiene está enriquecido en vesículas grandes.
A continuación, el sobrenadante se sometió a ultrafiltración para concentrar el volumen mediante el uso de un concentrador de 100 kDa tipo Amicon, y así tener un volumen adecuado para realizar el siguiente paso del protocolo, que fue la separación por cromatografía de exclusión molecular (SEC).
Fig. 1. Esquema del protocolo de aislamiento/enriquecimiento de las Brain-DerivedEVs (bdEVs) y de su caracterización posterior.
RESUMEN DE ALGUNOS RESULTADOS.
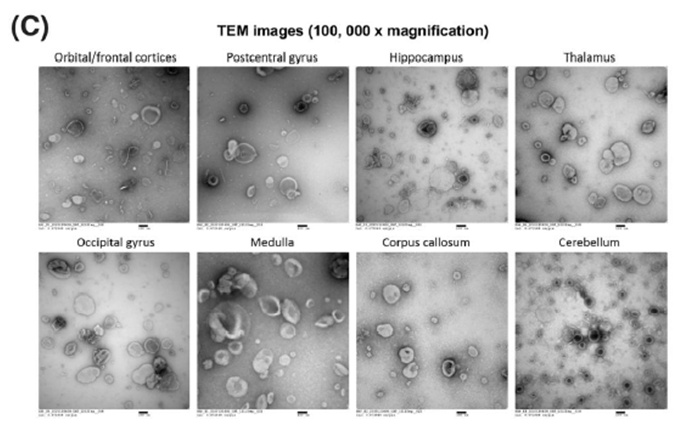
En este trabajo se caracterizaron las EVs de 8 regiones diferentes: giro orbitofrontal (ORB), giro post-central (POSTCENTRAL), hipotálamo (HIPPO), tálamo (THAL), giro occipital (OCC), médula (MED), cuerpo calloso (CORP) y cerebelo (CBLM).
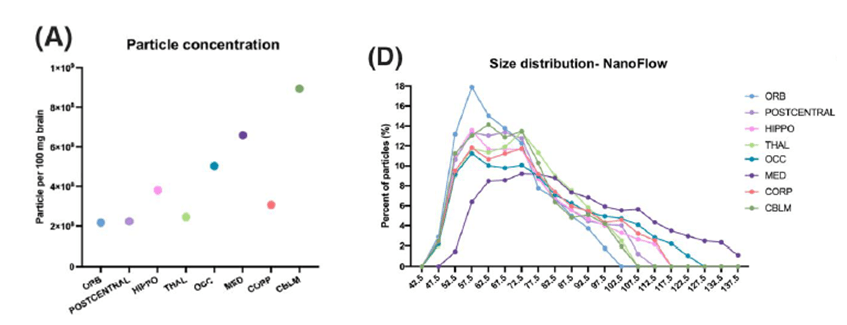
De todas estas regiones, la que mayor rendimiento tuvo en cuanto a cantidad de partículas por mg de tejido, fue el CBLM, mientras que el ORB tuvo una menor cantidad de bdEVs recuperadas (Fig.2A). En cuanto al tamaño de las bdEVs, los autores encontraron que las dbEVs derivadas de la médula eran las que mayor tamaño tenían (Fig. 2C y D), mientras que las dbEVs derivadas de ORB y CBLM eran las de menor tamaño.
Fig. 2. Concentración y tamaño de las bdEVs
Para analizar de qué tipos celularesprocedían las bdEVs purificadas, los autores analizaron diversos marcadores neuronales, microgliales, astrocitarios, comunes a todos los tipos celulares y marcadores no neurales mediante técnicas de ELISA (Fig.3).
Fig. 3. Análisis de la expresión de marcadores neurales en las bdEVs, mediante técnica de ELISA.
Se puede ver que, de entre los marcadores neurales, NCAM y CD90 son los que más se expresan en las bdEVs de todas las regiones analizadas, destacando un poco más NCAM en médula. Las bdEVs que expresan más CD271, denominado NGFR, se encuentran producidas en el CBLM.
En cuanto a marcadores astrocitarios, sólo CD44 se detectó en las bdEVs de las distintas regiones, destacando su expresión en la región OCC y en MED.
En este artículo también analizan la expresión de diferentes subtipos de RNA del interior de las bdEVs y determinan mediante análisis del componente principal (PCA) encuentran que existen perfiles de sRNA claramente diferentes entre CBLM, THAL y MED del resto bdEVs procedentes del resto de regiones analizadas.
CONCLUSIÓN
A pesar de que el artículo analizado tiene ciertas limitaciones, sobre todo en cuanto a la utilización de un único paciente como fuente de las bdEVs, que hace que los resultados obtenidos en cuanto a abundancia de bdEVs, expresión de marcadores o de RNAs no sean concluyentes, este trabajo proporciona un protocolo para obtener EVsa partir de un tejido que puede ser de gran utilidad en el futuro. Es necesaria la validación de estas observaciones en tejido de más pacientes, pero la metodología podrá permitirnos estudiar el origen y la posible función de las EVs producidas in situ.
AUTOR
Virginia Vila del Sol. Responsable del Servicio de Citometría y Unidad de Vesículas Extracelulares Hospital Nacional de Parapléjicos. Toledo, España.
Referencias
Y. Huang, T. Arab, A. E. Russell, E. R. Mallick, R. Nagaraj, E. Gizzie, J. Redding-Ochoa, J. C. Troncoso, O. Pletnikova, A. Turchinovich, D. A. Routenberg, K. W. Witwer, Interdiscip. Med. 2023, 1, e20230016. https://doi.org/10.1002/INMD.20230016
Para más información, dudas o solicitud de presupuestos puedes contactarnos en: uvex.hnp@sescam.jccm.es
En las dos Newsletter anteriores de esta serie, hemos descrito en detalle el microscopio IX83 de Olympus (Newsletter 1, de Enero 2022) y además hemos introducido los conceptos básicos de la inteligencia artificial directamente relacionados con el análisis de la imagen (Newsletter 2, de Enero 2023). En esta tercera y última Newsletter de la serie explicaremos tres ejemplos con los que hemos trabajado utilizando la IA para analizar las imágenes capturadas. Con los tres ejemplos que describimos podréis haceros una idea de la versatilidad que tenemos a la hora de analizar imágenes mediante IA.
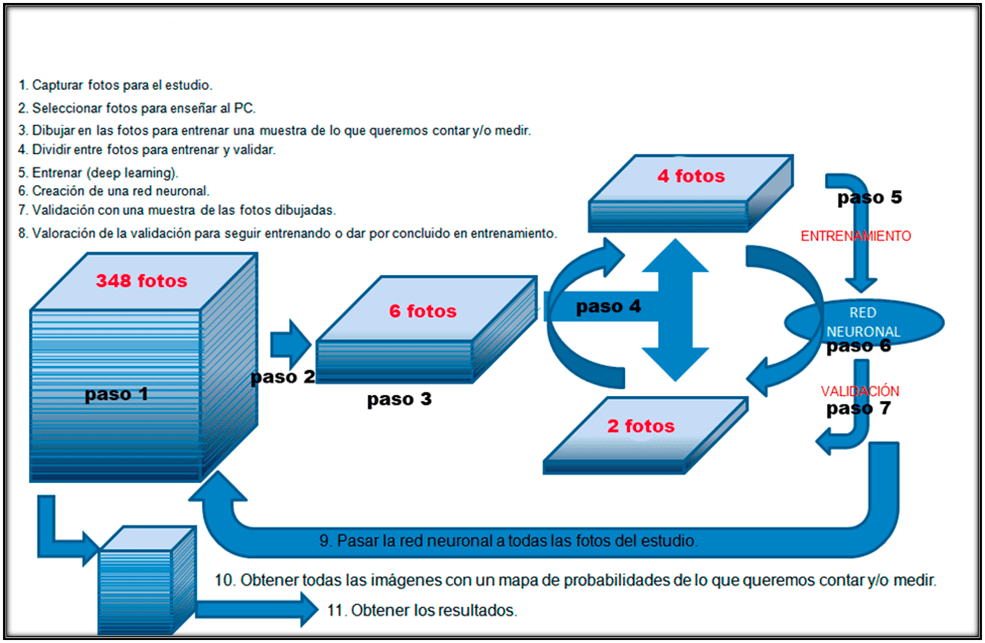
La Newsletter está estructurada en los diferentes pasos a seguir para realizar el proceso de Deep learning, que podéis ver resumidos en el esquema de la figura 1. Cada apartado de la Newsletter es uno de los pasos del esquema (figura1) esto permite seguir el proceso más fácilmente (pasos 1, paso 2, etc), y dentro de cada paso se explican los 3 ejemplos diferentes que estamos describiendo.
Nota: algunos pasos son simultáneos en el tiempo y se explican en un orden no consecutivo para entender mejor el proceso.
Figura 1. Esquema de los diferentes pasos en la utilización del Deep learning para crear redes neuronales para el análisis de imágenes mediante Inteligencia Artificial.
Los tres ejemplos que iremos explicando paso a paso son los siguientes:
EJEMPLO 1 Fotos fibras musculares. Fotos capturadas en un microscopio diferente al que se utilizara para crear la red neuronal. Identificación de fibras de musculo liso oxidativas y no oxidativas (tipo 1 y tipo 2) en diferentes animales con diferentes tratamientos.
EJEMPLO 2Fotos cortes de medula para contar neuronas. Fotos capturadas con el mismo equipo (microscopio IX83 de Olympus) y el mismo y software (Cellsense) con el que se entrena y crea la red neuronal con la que se analizarán las fotos capturadas. Identificación de neuronas en medula.
EJEMPLO 3 Fotos de células en cultivo para contar núcleos. Fotos capturadas con el mismo equipo (microscopio IX83 de Olympus) pero diferente software (ScanR) con el que se entrena y crea la red neuronal con la que se analizarán las fotos capturadas. Para identificación de núcleos raros (cuando los núcleos son poco convencionales debido a tratamientos especiales) para contar células en HCS (High Content Analisis).
A. CAPTURA
PASO 1. Captura de las fotos.
EJEMPLO 1. Fotos fibras musculares
Se utilizó el equipo de estereología (BX61), pero se podría haber utilizado cualquier microscopio de los que tenemos en el servicio, con una cámara color (DP-70) y un objetivo 10X, intentando hacer un máximo de 10 fotos por animal.
Figura 2. Algunas de las fotos capturadas de fibras musculares (Ejemplo 1)
EJEMPLO 2. Fotos cortes de médula espinal para contar neuronas. (Este sería el ejemplo más común de captura de fotos, el más automatizado y el que se realiza con el mismo microscopio donde se analizará).
1. Decisión las características del experimento de captura, principalmente tipo de iluminación (transmitida/ fluorescencia) potencia de la luz, filtros de fluorescencia, tiempo de exposición, offset etc. (Fijar Settings)
2. Determinar la ruta donde se almacenaran las fotos que capturaremos de forma automática y además el nombre* con el que se guardaran cada una de ellas.
Nombre*; nombre del experimento (general para todas las fotos)_separador_ posición de grupo (especifico para cada porta)_ separador_ contador (un número, especifico de cada corte o ROI)
3. A continuación se obtiene un mapa o vista general a baja resolución (4X) de la zona de la platina donde colocamos la muestra. Este mapa o vista general (overview) nos permitirá navegar por la muestra que tenemos en la platina. Con esto obtenemos una imagen a baja resolución de los portaobjetos colocados en el adaptador múltiple de la platina (de 1 a 4 porta objetos).
4. Determinar sobre el mapa o la vista general la ROI o zona de interés* que queremos fotografiar con mayor resolución (ver ROIs en azul figura 3).
5. Agrupamos cada grupo de cortes en otra ROI más grande (rojo, verde figura 3) que engloba todos los cortes de un mismo grupo (porta, tratamiento, individuo…), será lo que en el nombre definitivo de la foto se denomina posición de grupo (ver nombre*).
6. Fijación del mapa de foco*.Determinar tres puntos de foco por cada corte (ROI), para crear un plano de foco. De esta, durante la captura, el microscopio enfocará de manera automática a lo largo de la muestra que va capturando. Si la región a fotografiar está compuesta por 4 o menos campos podemos enfocar cada vez que se dibuja una ROI (azul figura 3) y no será necesario hacer el plano de foco.
*Mapa de foco; Fijar de 3 puntos de foco por cada región (ROI) a fotografiar, con tres puntos de foco definimos completamente el plano y por tanto se irá ajustando el foco a lo largo del plano a fotografiar en relación a los tres puntos de foco.
7. Capturamos a mayor resolución (10X en este ejemplo) las imágenes en mosaico de las regiones definidas anteriormente en el overview o mapa. En otros ejemplos se ha utilizado el objetivo 20X o el 40X (pero en este caso haciendo Z-stack y un proceso de EFI “Extended focus imaging”, para tener todos los planos en uno solo completamente enfocado).
En este ejemplo se diseña un experimento de captura de fotos en placas de cultivo utilizando el programa ScanR de Olympus. Este programa también lo tenemos en el microscopio IX83 motorizado de Olympus y está diseñado principalmente para hacer HCA (High Content Analysis), normalmente en placa de cultivo y captura de forma automática multitud de fotos de cada pocillo de la placa de cultivo.
El ScanR está compuesto por dos Softwares, el programa de Captura (para adaptar la captura a cualquier placa de cultivo) y el programa de Análisis (que es parecido a los programas de citometría, para analizar poblaciones de eventos). Con el ScanR podemos entrenar el equipo para crear redes neuronales que nos permitan analizar las imágenes aplicando inteligencia artificial (Newsletter de Jose Angel Rodriguez el 28 de Junio 2021). Pero en este caso, la red neuronal utilizada para el análisis de las fotos capturadas con ScanR la crearemos con el Software Cellsens. Esto lo hacemos puesto que el software Cellsens es más flexible a la hora de etiquetar las fotografías a analizar durante el entrenamiento (ver paso 3). Pese a que la captura e incluso el análisis se realizaran con otro programa (ScanR) podemos utilizar las fotos capturadas para crear una red neuronal en el programa Cellsens.
En este caso crearemos una red neuronal que identifica núcleos complejos (poco convencionales) y nucleos normales, para identificar células independientes en el proceso de HCA del ScnaR (ver Newsletter de Jose Angel Rodriguez el 28 de Junio 2021). Es decir, después de capturar las imágenes de los diferentes pocillos de la placa con ScanR de captura, utilizaremos algunas fotos para entrenar una red neuronal con Cellsens. Una vez tengamos la red neuronal, la utilizaremos en ScanR de análisis, para analizar y agrupar en poblaciones las células con núcleos raros de las fotografías capturadas en los diferentes pocillos de la placa de cultivos.
RESUMEN CAPTURA
Terminado el proceso de captura (Paso 1) tenemos multitud de fotos para analizar. Las fotografías que tenemos son:
Figura 3. Tipos y número de de fotos capturadas para cada tipo de ejemplo.
B. ENTRENAMIENTO
PASO·2. Selección de las fotos para entrenar
Una vez capturadas las fotos de cada ejemplo se realiza una selección al azar de entre un 1% a un 10% de las fotos capturadas (dependiendo del número total de fotos y de la complejidad de lo que queremos identificar), para realizar el entrenamiento de la red neuronal.
Ejemplo 1. De las fibras musculares se seleccionó un 10% de las fotos, en este caso seleccionamos un alto porcentaje de fotos para entrenar, debido a que el entrenamiento es un tanto complejo, puesto que debemos distinguir entre dos tipos de fibras y lo que no es fibra. Además no tenemos muchas fotos, por tanto un 10% no es un número excesivo para entrenar.
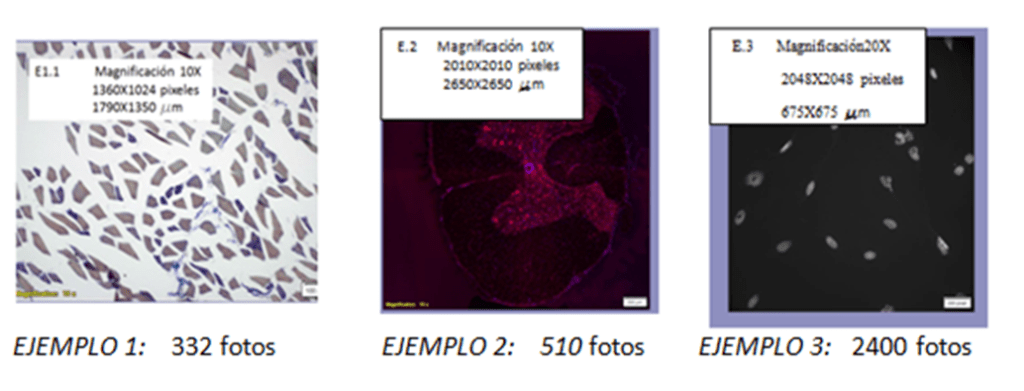
Se seleccionaron 33 fotos para entrenar de las 332 adquiridas.
Ejemplo 2. De los cortes de medula para la identificación de neuronas, se seleccionó un 5% de las fotos, en este caso las fotos son muchos más grandes y con más ejemplos de neuronas en cada una, además tenemos más fotos totales. En este ejemplo el problema es asegurarnos de introducir en la selección, fotos con lesión donde la zona de inflamación puede producir errores de contaje de neuronas, esto quiere decir que en este caso no debemos seleccionar todas las fotos al azar.
Se seleccionaron 25 fotos para entrenar de las 510 adquiridas.
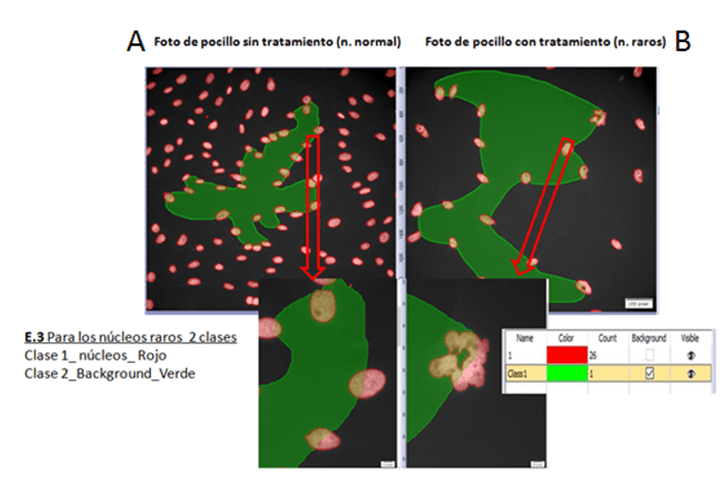
Ejemplo 3. De las fotos de núcleos raros seleccionamos sólo un 2 %. En este caso un 2% es un número elevado de fotos, puesto que partimos de 2400 fotos (se capturaron de forma automática con ScnaR). Aquí tenemos que asegurarnos de introducir fotos de pocillos sin tratamiento (con núcleos normales) y otras con núcleos raros (fotos de pocillos con tratamiento), para entrenar al PC en la identificación tanto de núcleos normales como lobulados o raros.
Se seleccionaron 48 fotos de entrenamiento (20 con núcleos normales y 28 con núcleos raros) de las 2400 adquiridas.
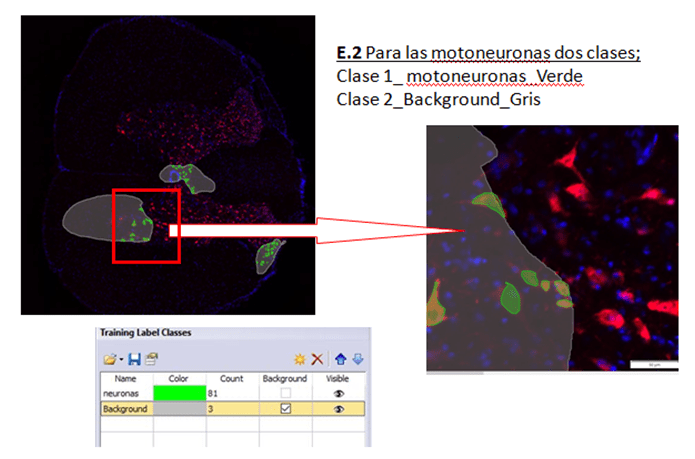
PASO 3. Etiquetado de imágenes. Dibujando sobre las fotos seleccionadas las mascaras que distingan las diferentes zonas o estructuras a identificar para el entrenamiento.
Una vez seleccionadas las fotos para el entrenamiento deben ser etiquetadas para identificar los objetos de interés para nuestro estudio. Para ello utilizamos la interface (pestaña o layout) de “Count and measure” del programa Cellsense (figura 4, 5 y 6).
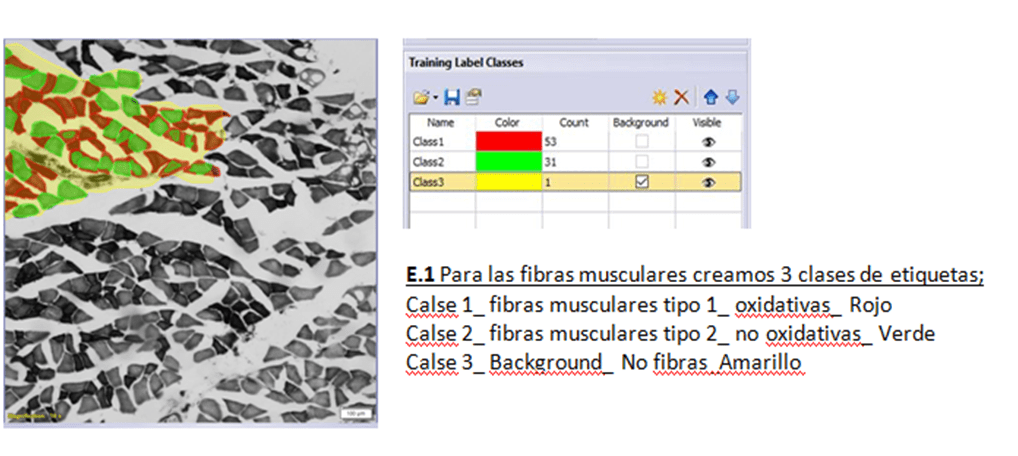
EJEMPLO 1
Figura 4. Etiquetas de entrenamiento para el ejemplo 1
EJEMPLO 2
Figura 5. Etiquetas de entrenamiento para el ejemplo 2
EJEMPLO 3
Figura 6. Etiquetas de entrenamiento para el ejemplo 3
Utilizando las etiquetas que se muestran en las figuras 4, 5 y 6, se dibujan manualmente por el investigador las fotos seleccionadas para entrenar. No es necesario etiquetar el 100 % de la foto y siempre es necesario crear la etiqueta de Background, donde se identifica todo lo que no interesa y que rodea a la zona etiquetada.
En el caso del ejemplo 3 el etiquetado fue semiautomático, para dibujar las máscaras que identifican los núcleos en las fotos para el entrenamiento, se recurrió a los pasos clásicos de segmentación de la imagen, mediante un ajuste de Threshold, un watershed y una binarización de forma automática. Tras obtener el resultado de segmentación, las fotos fueron editadas. Esta edición fue manual y consistió en ir uniendo zonas que el watershed había separado y separando zonas que el watershed no ha identificado, debido a la complejidad de los núcleos raros que tenían formas muy heterogéneas (ver Figura6 B). Esta parte de edición la hacemos de manera similar y con las mismas herramientas que utilizamos para dibujar las mascaras en los ejemplos 1 y 2.
C. ENTRENAMIENTO
PASO 5. Entrenamiento (Deep Learning). Nota: Pasos 4 y 5 son simultáneos en el tiempo (ver figura 1).
Para realizar el entrenamiento con el software Cellsens utilizamos el Layout o interface de Deep learning del programa (figura 7). En esta interface del programa añadimos las fotos etiquetadas o dibujadas en el paso anterior. Estas fotos y sus máscaras servirán para que el programa diseñe una red neuronal aprendiendo de las fotos marcadas o etiquetadas mediante deep learning (Newsletter 1, de Enero de 2022 o Newsletter 2, de Enero 2023).
Uno de los puntos importantes del entrenamiento es decidir el tipo de Deep learning que realizará el equipo y el número de iteraciones que realizará. Cada tipo de deep learning utiliza unos algoritmos diferentes, en los tres ejemplos que nos ocupan, hemos utilizado el estándar (Standard Network). Por último, se fijan el número de iteraciones que realizara el entrenamiento. Las iteraciones son repeticiones del proceso de entrenamiento hasta obtener el resultado deseado, comenzamos con 250 k pero podemos llegar a más de un millón.
Nota: Standard Network, utiliza una arquitectura U-Net con 32 mapas de características en la primera capa de convolución. La red aplica rotaciones de 90° y duplicación de las imágenes etiquetadas para aumentar los datos de entrenamiento.
PASO 4. Selección de las imágenes de validación. Nota: Pasos 4 y 5 son simultáneos en el tiempo (ver figura 1).
Este paso es automático, y lo realiza el propio programa una vez comience el proceso de entrenamiento. En el paso anterior son cargadas las fotos etiquetadas en la ventana de entrenamiento. Y de todas las fotos que fueron etiquetadas manualmente y cargadas en la interface de entrenamiento, el programa selecciona de forma automática entre un 1 y el 10 % de estas fotos, y las utilizara para validar, sin ser utilizadas para el entrenamiento. Es decir, de las fotos etiquetadas manualmente, el programa separa un pequeño porcentaje de ellas que serán utilizadas para validar y no para entrenar.
E1. Fotos adquiridas: 332. Fotos de entrenamiento: 33. Fotos de validación: 3
E2. Fotos adquiridas: 510. Fotos de entrenamiento: 25. Fotos de validación: 3
E3. Fotos adquiridas: 2400. Fotos de entrenamiento: 48. Fotos de validación: 5
D. VALIDACIÓN.
PASO 7 y 8. Validación y valoración de la Validación. Nota: Pasos 6, 7 y 8 son simultáneos en el tiempo (ver figura 1).
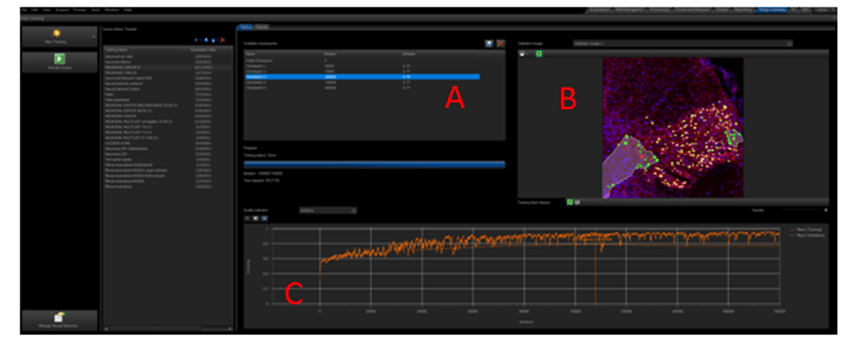
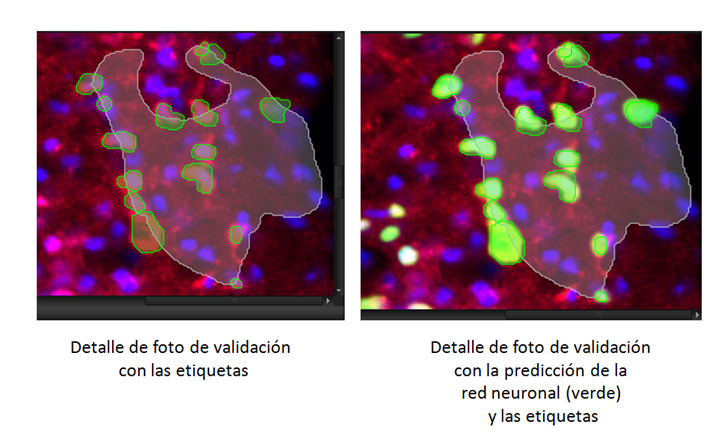
Cuando comienza el entrenamiento, el programa muestra una grafica en la que se representan valores de similaridad frente a las iteraciones realizadas durante el entrenamiento (figura7 C), cuantas más iteraciones se produzcan mayor será la similaridad, hasta un punto en el que el valor se mantiene constante o incluso empieza a decaer. Pero, ¿cómo podemos definir el valor de similaridad? Pues bien, el valor de similaridad es, como se parece la predicción hecha por el programa aplicando la red neuronal creada hasta ese momento (después de realizar unas iteraciones concretas) con las etiquetas de las fotos de validación que hemos dibujado manualmente en las fotografías seleccionadas para validar (es decir, en las fotografías etiquetadas y reservadas para validación sin ser utilizadas para crear la red neuronal). Los valores de similaridad son un porcentaje y se representan de 0 a 1. Es importante resaltar que las fotos de validación no se utilizaron en el entrenamiento para crear la red neuronal. De manera que pese a estar etiquetadas el programa no las ha utilizado para aprender. Además cada cierto número de iteraciones el programa ira fijando automáticamente unos check-point, con los valores de similaridad específicos a esas iteraciones.
Figura 7. Layout o interface de Deep learning en el software Cellsens. Donde vemos los diferentes check point (A), una imagen de validación (B) y la grafica de similitud con respecto a las iteraciones (C).
Y aquí tenemos la grafica de validación (Figura 7C) y la imagen de validación (Figura 7B y Figura 8) con la predicción hecha sobre la imagen a las 110000 iteraciones o Check point 3 (Figura 7A).
Figura 8. Detalle de la foto de validación al comienzo del entrenamiento y en el tercer check point (110000 iteraciones) con un valor alto de similaridad.
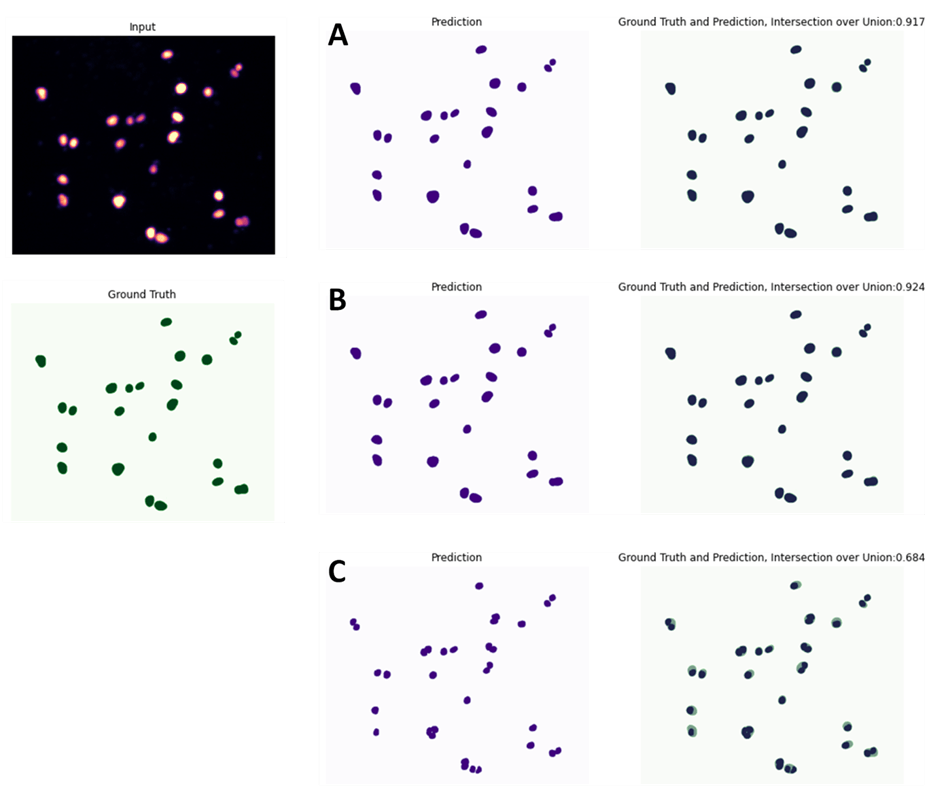
Cuando los valores de validación o similaridad son altos (cercanos al 0.8, es decir un 80 % de similaridad), es porque la predicción de la red neuronal creada hasta el momento considera que se aproxima mucho a las etiquetas que creamos manualmente. Existen diferentes medidas de validación, las más comunes son; el valor de similaridad, el Intersection over Union (IoU) o el coeficiente de similitud. Estos valores sirven para evaluar los algoritmos de Deep Learning al estimar como de bien una máscara predicha coincide con los datos reales del terreno. Es obvio, que cuanto mayor sea el valor de similaridad más se aproxima la predicción a la realidad. Así podemos ver que cerca de 110000 iteraciones del entrenamiento de las neuronas (E.2) tenemos unos valores de similaridad de más del 0.8 y que ese valor no aumenta por mas iteraciones que se realicen. Lo cual nos lleva a decidir que la red neuronal está terminada y podemos finalizar el proceso de entrenamiento fijando un check point a este número de iteraciones (ver figura 7).
E.1. En el caso del musculo programamos 150000 iteraciones y vimos que a partir de 30000 iteraciones los valores de similaridad eran altos llegando al máximo a las 50000 iteraciones con un Intersection over Union (IoU) de 0.79, a partir de aquí se estanco y a las 100000 iteraciones empezó a bajar el Intersection over Union (IoU).
E.3. En el caso de los núcleos se programaron 100000 iteraciones y en el primer check point de 20000 iteraciones el valor de similaridad ya era del 0.94.
E. RED NEURONAL CREADA DURANTE EL ENTRENAMIENTO.
PASO 6. Obtención de la red neuronal Nota: Pasos 6, 7 y 8 son simultáneos en el tiempo (ver figura 1).
Durante todo el proceso de entrenamiento mediante Deep learning el programa ha ido gravando automáticamente unos puntos de comprobación o Check point (figura 7 A). Esto lo se ha hecho de forma automática cada cierto tiempo, o mejor dicho, cada cierto número de iteraciones. Estos check-point también han podido ser grabados bajo nuestra supervisión a ciertas iteraciones en las que nos pareciera que los valores de similaridad eran suficientemente altos.
Con todos estos puntos de comprobación o check-point decidimos cual es el mejor para nuestro propósito y lo guardamos como red neuronal.
Una vez que tenemos la red neuronal, podremos identificar por Inteligencia Artificial las fibras musculares tipo 1 o 2 (ejemplo 1), las neuronas (ejemplo 2) o los núcleos aunque estos sean raros (ejemplo 3).
F. OBTENCIÓN DEL MAPA DE PROBABILIDADES Y RESULTADOS.
PASO 9. Utilizar la red neuronal para identificar el propósito de nuestro estudio en el total de las fotos, obteniendo un mapa de probabilidad (PASO 10) y a partir del mapa de probabilidad los resultados (PASO 11).
Con la red neuronal establecida, se hace un análisis de todas las fotos del estudio aplicando la red neuronal a todas las fotos capturadas del experimento. Tras aplicar la red neuronal obtenemos una capa adicional sobre la foto capturada, que representa un mapa de probabilidades de identificación de las estructuras etiquetadas para el entrenamiento. El mapa de probabilidades representa la probabilidad de cada pixel de la foto a pertenecer a cada una de las clases etiquetadas en el entrenamiento A partir del mapa de probabilidad podemos obtener los resultados de diferentes formas.
E1. MÚSCULO. Utilizando la interface de “count and measure” del programa Cellsens creamos una macro con la pestaña “macro manager”. Esta macro consiste en aplicar la red neuronal obtenida en el paso anterior a todas y cada una de las fotos adquiridas, para poder hacer el proceso automáticamente (esta parte es común a los tres ejemplos).
Resultado de esta macro obtenemos una capa adicional en la imagen capturada que representa un mapa de probabilidades por cada pixel de la foto, que nos indica la probabilidad a pertenecer a cada una de las clases etiquetadas en el entrenamiento, es decir, a cada una de las fibras musculares o al background (figura 9 B).
Con el mapa de probabilidades por tipos de fibra creamos una segunda macro, para segmentar la imagen del mapa de probabilidad mediante un threshold. Determinamos un Threshold (umbral) para cada tipo de fibra y para el background y corremos la macro en las fotos con el mapa de probabilidades. De esta manera cuando corremos la macro obtendremos una imagen binarizada por tipo de fibra y background y una tabla con el porcentaje y área de cada tipo de fibra por foto (Figura 9C).
Figura 9. Detalle de una foto de fibras musculares (Ejemplo.1)( A), la misma foto con su mapa de probabilidad (B) y la misma imagen binarizada tras la segmentación que dará lugar a los resultados (C).
E2. NEURONAS. Pasamos la red neuronal obteniendo los mapas de probabilidad de cada una de las fotos. A partir de estas imágenes se obtuvieron los mapas de probabilidad como en el caso anterior. Se separaron las capas de los mapas de probabilidad y se analizaron en el programa libre Fiji. A partir de los mapas de probabilidad aislados, se segmentaron de forma muy sencilla y se contaron mediante la función de análisis de partículas de Fiji, obteniendo el número de neuronas por cada foto (corte).
E3. NUCLEOS RAROS. Una tercera opción que podemos hacer es enviar la red neuronal a el programa ScnaR y utilizarla para analizar las fotos capturadas para hacer HCA, en este caso para identificar los núcleos (sean normales o raros) con esta red neuronal. Utilizando la red neuronal para segmentar las imágenes capturadas con ScanR.
AUTOR José Ángel Rodríguez Alfaro Responsable del Servicio de Microscopía y Análisis de Imagen Hospital Nacional de Parapléjicos. Toledo, España.
En una de nuestras primeras newsletters, os hablábamos sobre la metodología SWATH-MS (Sequential Window Acquisition of All Theoretical Fragment Ion Mass Spectra). Este tipo de tecnología, de adquisición independiente de datos (DIA) de espectros en espectrometría de masas, en estos años se ha revelado como una de las más prometedoras para mejorar la cobertura y la cuantificación de proteínas en mezclas complejas.
En esta newsletter vamos a profundizar un poco más sobre las estrategias y aplicaciones de este enfoque en análisis “ómicos”
¿Qué es una adquisición independiente de datos de espectros (DIA)?
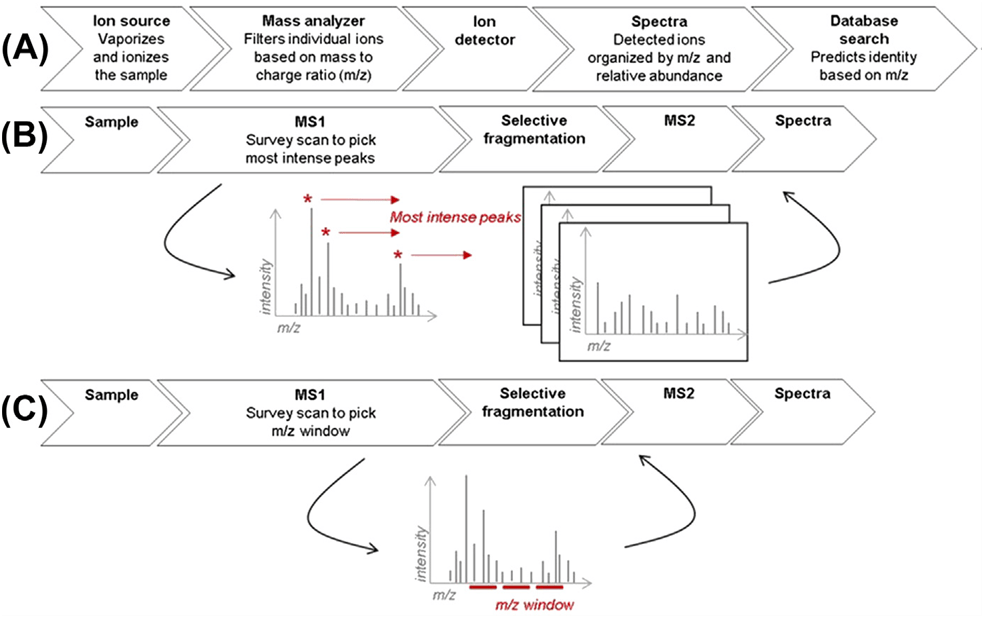
El propio nombre lo indica. Es un método de adquisición con el que el espectrómetro nos va a recoger todos los datos de espectros de la muestra. Para un método de adquisición dependiente de datos el espectrómetro va a seleccionar únicamente aquellos iones que cumplan con unos criterios previos establecidos, mientras que en los métodos DIA, el espectrómetro va analizando por ventanas todos los iones que encuentre (Figura 1).
Figura 1: Espectrometría de masas en tándem para proteómica cuantitativa. (A) Esquema básico de espectrometría de masas. (B) Adquisición dependiente de datos. Los iones precursores se seleccionan en función de un umbral predefinido. (C) Adquisición independiente de datos. Los iones precursores se seleccionan en una ventana m/z.
¿Por qué SWATH-MS?
SWATH-MS es como ha denominado Sciex a los métodos DIA de sus equiposzenoTOF, QTOF y TripleTOF de escaneo rápido, en los que se adquiere en ventanas de forma secuencial todos los fragmentos de masas de los iones que se registran previamente. Estas “ventanas” son intervalos de m/z que pueden ser de un ancho fijo o variables.
La ventaja que presenta tomar ventanas de tamaño variable es que podemos seleccionar intervalos más estrechos en zonas de nuestra cromatografía donde haya una mayor densidad de los precursores de analitos. Así, se logra una mejor especificidad en matrices complejas, ecualizando la densidad de precursores en cada una de las ventanas de aislamiento (Figura 2).
Figura 2: Investigación de anchos de ventana variables del Q1 para la adquisición de SWATH. Los histogramas de densidad m/z construidos a partir de los datos TOF MS para la muestra de interés (línea azul) se pueden usar para construir un patrón de ventana de tamaño variable (línea roja) usando la calculadora de ventana variable SWATH.3 En la figura se muestra una distribución m/z típica y un patrón de ventana Q1 utilizado para muestras de proteoma digeridas.
Cada casa comercial ha intentado implementar este tipo de adquisición en sus instrumentos con rendimientos más o menos limitadospara la interpretación de matrices complejas. Para conseguir explotar al máximo los datos obtenidos, se necesitan herramientas informáticas que nos ayuden en el análisis.
El desafío analítico que presenta la interpretación de datos DIA es la distinción de múltiples péptidos que están coaislados y cofragmentados en la misma ventana de selección de precursores. Es decir, perdemos esa relación directa que teníamos en ensayos DDA del precursor con sus iones fragmentos.
Estrategias de análisis
Para la interpretación de datos DIA podemos ayudarnos de software basados en:
Utilización de bibliotecas espectrales.
Sin bibliotecas espectrales.
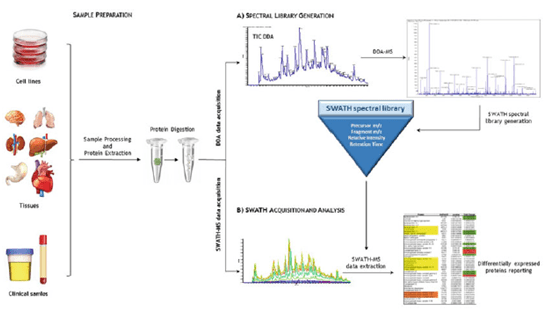
Figura 3: Representación esquemática de las estrategias para construir la biblioteca espectral en el análisis SWATH-MS. (A) La biblioteca espectral se obtiene de un experimento DDA simple. Contiene toda la información sobre un péptido identificado determinado (como tR, precursor m/z y espectros MS/MS). (B) Una vez realizada la biblioteca, las muestras se ejecutan en modo SWATH
Utilización de bibliotecas espectrales.
De forma sencilla, una biblioteca espectral generalmente se compone de un conjunto de coordenadas que incluyen el precursor m/z, el ion fragmentado m/z, la intensidad relativa de los iones fragmentados y el tiempo de retención estandarizado (RT) para cada precursor peptídico y sus fragmentos. La biblioteca espectral se utiliza para extraer las cromatografías iónicas de los péptidos objetivos de los espectros DIA.
El éxito de los experimentos DIA depende de la calidad de las bibliotecas espectrales utilizadas para la búsqueda en la base de datos. Con frecuencia, estas bibliotecas deben generarse mediante experimentos de adquisición dependiente de datos (DDA) fraccionados de las mismas muestras con las que se va a realizar el ensayo DIA. Esto que requieren mucho tiempo y trabajo.
¿Cómo construimos una biblioteca espectral? En teoría, todas las herramientas de software de búsqueda de espectro MS/MS pueden usarse potencialmente para construir las bibliotecas DIA.
Podemos resumirlo en tres pasos: i) recopilación de espectros de péptidos identificados con confianza en los motores de búsqueda, ii) integración de espectros del mismo ion peptídico en un único espectro representativo y tiempo de retención normalizado, iii) control de calidad para eliminar espectros no confiables Recientemente se han publicado varios algoritmos que permiten generar bibliotecas teóricas mediante una predicción eficaz del tiempo de retención y la intensidad de los iones fragmento.
Para generar bibliotecas teóricas fiables que puedan utilizarse en experimentos SWATH, se han desarrolladoherramientas de aprendizaje profundo para el análisis SWATH, con el fin de mejorar la sensibilidad y especificidad de los datos generados por espectrómetros de masas Q-TOF.
Sin bibliotecas espectrales.
En principio, si un péptido no está presente en la biblioteca espectral, no podrá analizarse utilizando métodos basados en bibliotecas. Así, se han desarrollado alternativas a los análisis DIA denominados “libres de bibliotecas” o método centrado en el espectro.
Aunque la búsqueda sin bibliotecas centrada en el espectro ha demostrado su potencial para identificar péptidos nuevos, la mayoría de los estudios publicados todavía utilizan el enfoque de extracción dirigida basado en bibliotecas espectrales.
Aplicaciones.
Bien, después de este despliegue de información sobre qué es un análisis DIA y sus estrategias de análisis, ¿en qué aplicaciones según las necesidades de mi trabajo de investigación puedo considerarlo?
Si el objetivo de nuestro proyecto es identificar el número máximo de proteínas a partir de un número limitado de muestras de una determinada complejidad, DIA no debería ser nuestro método de elección. Para este tipo de estudios la metodología basada en fraccionamiento de péptidos combinada con gradientes largos y métodos DDA tienen una mayor profundidad en identificaciones.Aunque tengamos una limitación con la cantidad de muestra necesaria para la realización del ensayo.
En los enfoques DIA, las concentraciones requeridas son menores (0.1-1µg) y la principal ventaja es poder utilizar gradientes de cromatografía cortos para poder analizar un proteoma tisular completo
DIA-MS está diseñado para combinar el potencial de la metodología no dirigida (discoveryshotgun, DDA) y el enfoque dirigido (SRM/MRM).En concreto, SWATH-MS se presentó como una alternativa para superar las limitaciones de métodos de cuantificación sin etiqueta. Al tratarse de un método insesgado, capaz de cuantificar un gran número de péptidos con una consistencia y precisión similares a las de métodos dirigidos como MRM, lo que convierte en una estrategia prometedora para el descubrimiento de biomarcadores a partir de cribados a gran escala.
Cuando se evalúa la opción de enfoques DIA, no solo debe considerar la identificación objetiva de proteínas y la precisión cuantitativa, que se han realizado para los datos SWATH, sino que también debe tener en cuenta otros factores cruciales, incluido el objetivo de la investigación, la estrategia de adquisición de datos DIA, rendimiento, consumo de cantidad de muestra, instrumentos de MS, viabilidad y costo del análisis.
Referencias aplicaciones
Klont F, Jahn S, Grivet C, König S, Bonner R, Hopfgartner G. SWATH data independent acquisition mass spectrometry for screening of xenobiotics in biological fluids: Opportunities and challenges for data processing. Talanta. 2020 May1;211:120747. doi: 10.1016/j.talanta.2020.120747. Epub 2020 Jan 15. PMID: 32070597.
Anjo, S.I.; Santa, C.; Manadas, B. SWATH-MS as a Tool for Biomarker Discovery: From Basic Research to Clinical Applications.Proteomics2017,17doi.org/10.1002/pmic.201600278
Finamore F, Cecchettini A, Ceccherini E, Signore G, Ferro F, Rocchiccioli S, Baldini C. Characterization of Extracellular Vesicle Cargo in Sjögren’s Syndrome through a SWATH-MS Proteomics Approach. International Journal of Molecular Sciences. 2021; 22(9):4864. https://www.mdpi.com/1422-0067/22/9/4864
Hallal S, Azimi A, Wei H, Ho N, Lee MYT, Sim H-W, Sy J, Shivalingam B, Buckland ME, Alexander-Kaufman KL. A Comprehensive Proteomic SWATH-MS Workflow for Profiling Blood Extracellular Vesicles: A New Avenue for Glioma Tumour Surveillance.International Journal of Molecular Sciences. 2020; 21(13):4754. https://www.mdpi.com/1422-0067/21/13/4754
Turner N, Abeysinghe P, Flay H, Meier S, Sadowski P, Mitchell MD. SWATH-MS Analysis of Blood Plasma and Circulating Small Extracellular Vesicles Enables Detection of Putative Protein Biomarkers of Fertility in Young and Aged Dairy Cows Journal of Proteome Research 2023 doi.org/10.1021/acs.jproteome.3c00406
AUTOR
Gemma Barroso García, MSc. Responsble SAI-Proteómica. Hospital Nacional de Parapléjicos. Toledo, España.
Para más información, dudas o solicitud de presupuestos puedes contactarnos en: unidadproteomica.hnp@sescam.jccm.es
Es una herramienta genética que permite eliminar o activar la función de cualquier gen en las diferentes células de un ratón, organismo modelo por excelencia. Se utiliza para crear diferentes de modelos de ratón: knockouts específicos de tejido, knockouts generales, knockouts inducibles y encender genes fluorescentes o informadores.
¿QUÉ ES EL CRE-LOX?
Este sistema se tomó del mecanismo de recombinación del Bacteriófago P1, el cual inserta y retira su genoma del genoma de la bacteria mediante el uso de la recombinasa «Cre» que reconoce a las secuencias de nucleótidos que reciben el nombre de sitios LoxP
Durante el ciclo de vida viral del bacteriófago, el fago se adhiere al exterior de la pared bacteriana y luego, una vez adherido, inyecta su ADN en el citoplasma de la bacteria. A continuación, el ADN del fago debe circularizarse para que el huésped lo replique.
Este bacteriófago tiene codificado en su propio genoma las enzimas necesarias para circularizar su ADN, así como secuencias diana de recombinación específicas. Este sistema natural tiene la capacidad única para hacer cortes específicos en el ADN y ligar el ADN de nuevo.
VENTAJAS DEL SISTEMA CRE-LOX
Permite a los investigadores enfocarse en porciones del genoma de forma controlable, en un lugar específico y en un momento específico. Esta tecnología permite supresiones de ADN muy específicas y dirigidas al mismo tiempo, lo que permite regular cuándo o dónde se elimina un gen.
También puede permitir a los investigadores la capacidad de evaluar la función de un gen en un tipo de tejido específico.
SISTEMA CRE-LOX
El sistema Cre-lox tiene dos componentes principales: la recombinasaCre y su secuencia objetivo, los sitios loxP. La Crerecombinasa es una enzima que genera roturas de ADN de doble cadena dentro secuencias específicas y liga las hebras de nuevo.
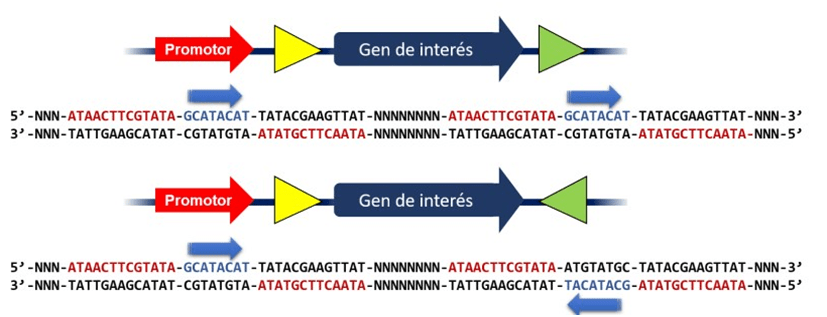
Las secuencias de ADN que se reconocen se denominan sitios loxP. Un sitio loxP tiene 34 pares de bases de largo y consta de dos repeticiones invertidas de 13 pb que flanquean una secuencia central de 8 pares de bases.
Dos moléculas de recombinasaCre se unen a cada una de las secuencias repetidas.
MECANISMOS CRE-LOX
Dependiendo de cómo estén orientados los sitios loxP en el genoma, el sistema Cre-lox puede servir para generar una delección (ambos orientados en la misma dirección) o una inversión (orientados en diferente dirección). El sentido lo otorga la secuencia interna azul flanqueada por las secuencias rojas invertidas.
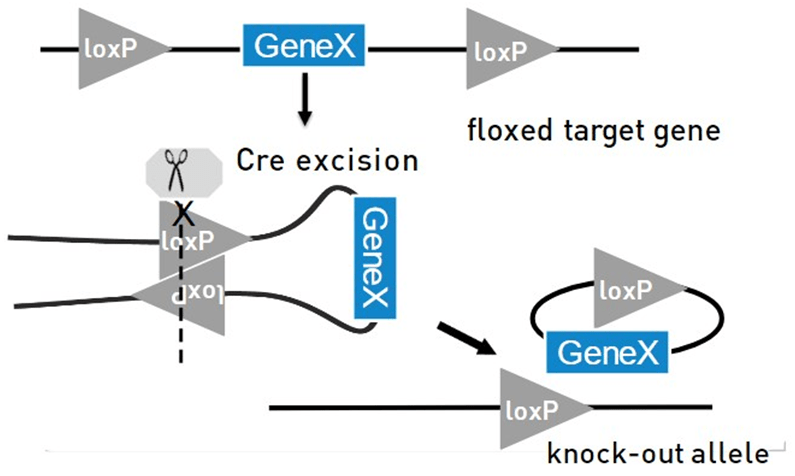
Mecanismo básico de Cre-lox para la eliminación de genes se debe insertar dos sitios loxP que flanquean la región a eliminar. Dependiendo de dónde se inserten y su orientación se pueden obtener diferentes tipos de eliminaciones.
Por lo general, se insertan estos sitios utilizando técnicas de recombinación homóloga a partir de células madre embrionarias que luego se utilizan para crear un ratón. Pero también se podría usar la tecnología CRISPR para eliminar los pasos de células madre embrionarias y crear modelos en una variedad de fondos genéticos eliminando el gen en ovocitos fertilizados. Cuando inserta los dos sitios loxP dentro de un gen, comúnmente nos referimos a eso como gen floxed, que es la abreviatura de flanked by loxP.
En presencia de Cre recombinasa, Cre se unirá a cada sitio loxP formando un tetrámero, y las dos hebras de ADN se emparejarán. La recombinasa Cre entonces hace un corte a mitad de camino a través de ambos sitios de reconocimiento en la secuencia central, y luego, liga las dos mitades de los diferentes sitios juntos. Como resultado, generará un episoma que contiene la pieza de ADN que será eliminado (Knock-out)
El sistema Cre-lox también se puede utilizar para crear inversiones cromosómicas esto se logra colocando los sitios loxP en una orientación inversa.
Cuando los sitios loxP se encuentran de esta manera, al emparejarse el ADN y la recombinasa el ADN se corta de tal manera que no se une la secuencia flanqueada, sino que intercambia los extremos y voltea la secuencia. Esto implica que la secuencia del gen ya no está dentro del sentido del promotor lo que lleva al silenciamiento del gen pero sin perderlo.
Si se reintroduce la recombinasa el gen puede activarse nuevamente (Knockout, inducible y reversible)
Además, si el transgen Cre está bajo el control de un promotor tejido-específico, la inactivación del gen se presentaría únicamente en tejidos específicos (knock-out condicional), permitiendo ratones genéticamente modificados de manera endógena.
Debido a que muchos genes inactivados por knock-out pueden ser esenciales para el desarrollo embrionario, esta tecnología sirve para evitar el problema de las metodologías mencionadas anteriormente.
Por ejemplo, si se emplea un ratón donde el gen de la adrenomedulina está “floxeado”, cuando estos ratones se cruzan con otros que portan la proteína Cre controlada por un promotor específico para neuronas, la proteína Cre se expresará sólo en neuronas y en estas células se producirá la ablación del gen de la adrenomedulina. Sin embargo, en el resto de células no se producirá la ablación del gen a pesar de que en todas ellas el gen está “floxeado”, ya que en estas células no se expresa la proteína Cre. De esta forma se obtienen ratones que viven sin problemas, ya que tienen una expresión relativamente normal de adrenomedulina, pero cuyas neuronas carecen de este gen, permitiendo el estudio detallado de las funciones del gen de la adrenomedulina en el sistema nervioso (Lack of adrenomedullin in the mouse brain results in behavioral changes, anxiety, and lower survival under stress conditions. Fernandez et al., 2008).
Obviamente, una vez que se tiene una línea de ratones con un gen “floxeado”, ésta se puede cruzar con distintos transgénicos para conseguir la delección del gen en distintos órganos o tejidos.
Si el promotor que se emplea para controlar la expresión de Cre se activa en un momento concreto del ciclo vital del animal, tendremos un knockout controlado temporalmente. De forma similar, si el promotor es sensible a la presencia de fármacos tendremos un knockout inducible, de forma que podremos disparar la destrucción del gen en el momento en que inyectemos la sustancia adecuada. Por supuesto, cualquier combinación de todo lo anterior es posible y la imaginación del investigador es el único límite para este tipo de tecnología.
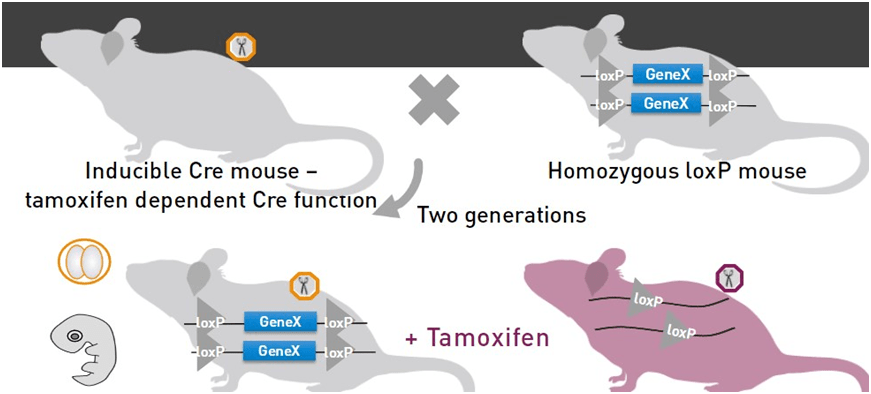
MODELOS DE RATÓN CRE INDUCIBLES
En el caso de los modelos inducibles lo que tenemos es un tipo de recombinasaCre que permanece inactiva hasta que introducimos un ligando o sustrato que le permita activarse. Estos tipos de sistemas Cre son útiles en situaciones en las que queremos controlar el momento de la actividad Cre, durante una etapa específica de desarrollo o edad.
El tipo más común de Cre inducible es un sistema inducible por tamoxifeno que permite que Cre se transloque al núcleo donde se activa y puede llevar a cabo el evento de eliminación y recombinación.
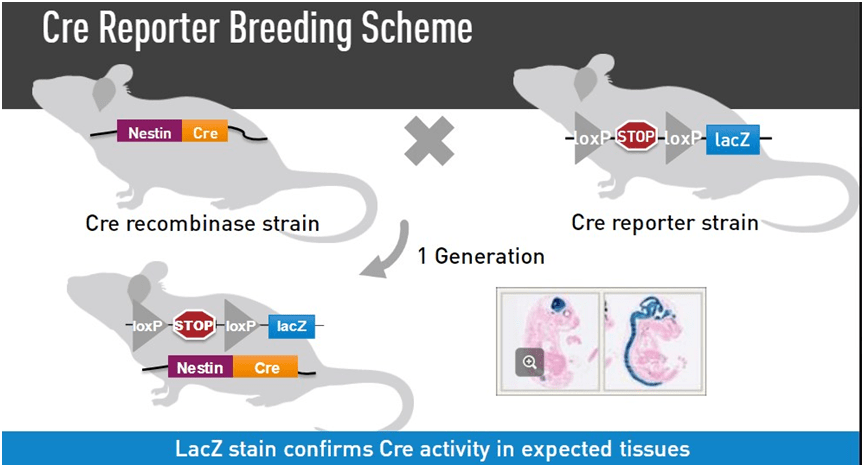
Una cepa reportera es una cepa en la que una proteína reportera, que suele ser algo que se puede detectar visualmente, como GFP o RFP, incluso lacZ, se expresa sólo cuando está en presencia de Cre.
La forma que se ha ideado para hacer esto es integrar un casete de parada transcripcional antes de la secuencia que codifica para el reportero (en este caso lacZ) y se flanquea el casete de parada con dos sitios loxP. Por lo tanto, en presencia de Cre, el casete de parada se elimina y el gen reportero puede ser expresado.
Este tipo de casetes LSL, o casetes lox-stop-lox con estos reporteros permiten a los investigadores evaluar la actividad de Cre usando la proteína fluorescente o lacZ como sustituto.
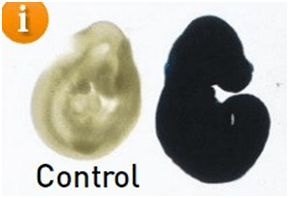
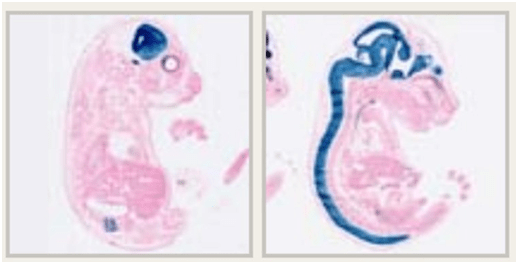
Un ejemplo de una cepa reportera Cre conlacZ-reporter se cruzó con una cepa Cre generalizada y la expresión de lacZ se detecta mediante tinción con Bgal.
El embrión control no tiene tinción, pero la cepa reportera cruzada muestra actividad generalizada.
Se puede utilizar esto para probar y evaluar cualquier cepa Cre específica.
Si se quiere ver la expresión de Cre en el sistema nervioso y tenemos una línea de NestinCre, se cruza con un reportero lacZ y se evalúa la actividad del reportero en las células de interés, además se comprueba si se expresa en otros lugares.
En este caso la actividad Cre es activa en el cerebro, la médula espinal y otras partes del sistema nervioso central y periférico de estos embriones E15.5, lo que confirma la especificidad que se está buscando.
El sistema Cre/lox es una de las herramientas más potentes y versátiles desarrolladas para la genética de ratones. Proporciona a los investigadores que utilizan ratones un control sofisticado sobre la ubicación y el momento de la expresión genética. Cre/lox se utiliza normalmente para producir alelos knockout, pero también se puede utilizar para activar la expresión genética.
El procesamiento de una muestra da lugar a una población de células muertas y debris celular, independientemente del protocolo empleado. La eliminación de dichas células del posterior análisis es un paso crítico en citometría de flujo, ya que pueden unir anticuerpo de manera inespecífica, dar autofluorescencia o incluso formar agregados al ser más pegajosas. A diferencia de la necrosis o muerte celular accidental, la apoptosis, o muerte celular programada, es un proceso muy controlado y complejo que se produce de forma natural en las células. A continuación veremos ejemplos de tintes con los que medir viabilidad celular así como ensayos de medida de apoptosis a través de alteraciones en la membrana plasmática o fragmentación de ADN mediante citometría de flujo.
ETIDIO Y CALCEÍNA
Mediante la tinción simultánea con Calceína AM y Etidio podemos discriminar rápidamente las células vivas de las muertas.
La Calceína AM posee una cola AM (acetoximetilo) unida por medio de un éster a la calceína, la cual le permite el paso por la membrana plasmática. Una vez dentro, las esterasas intracelulares rompen dicha cola reteniendo el colorante y haciendo que las célula vivas se tiñan de verde fluorescente (Calceína+Etidio-). El Etidio, se une al ADN y da un color rojo fluorescente para indicar la pérdida de integridad de la membrana plasmática en las células muertas (Calceína-Etidio+).
En el caso de las células moribundas, los poros en sus membranas hacen que la Calceína quede libre pudiendo salir de las células y dar lugar a una intensidad de fluorescencia menor. Además, el Etidio también será capaz de llegar al núcleo y unirse al ADN (Calceína+Etidio+).
Figura 1.Datos pertenecientes al grupo Fisiopatología Vascular (FPVI).
ANEXINA V Y 7AAD
Uno de los ensayos de apoptosis más importantes es la medida de alteraciones de membrana mediante ANEXINA V.
En las células vivas, los residuos de fosfatidilserina se encuentran en la cara interna de la membrana plasmática. Cuando la célula entra en apoptosis, se produce un cambio en la membrana que consiste en la externalización de dichos residuos.
La Anexina V es una proteína recombinante que se une específicamente a los residuos de fosfatidilserina cuando estos están expuestos en la cara externa de la membrana plasmática, siendo un biomarcador efectivo en células apoptóticas. La Anexina V puede ser combinada con un marcador de ADN como el 7-AAD o el yoduro de propidio (PI), con el fin de distinguir células apoptóticas de necróticas. Estos tintes no pueden atravesar la membrana celular. Sin embargo, en el caso de células muertas o moribundas, en las que la membrana plasmática presentan poros, estos tintes pueden penetrar fácilmente e intercalarse en el ADN celular generando una señal fluorescente específica. De manera que, las células que solo son AV+7AAD-, son células con apoptosis temprana; mientras que, las AV+7AAD+, serían células que sufren apoptosis tardía o muerte.
TUNEL (Terminal dUTP Nick End-Labeling)
Uno de los ensayos ampliamente empleados para la determinación y cuantificación de la apoptosis “in situ” es el denominado TUNEL. Este ensayo permite la determinación de los fragmentos de ADN cortados por la acción de endonucleasas activadas por caspasas, familia de proteínas clave en la ejecución de la muerte celular programada.
Los extremos 3’-OH del ADN dañado se marcan fluorescentemente mediante la adición de dUTP nucleótidos catalizada por la enzima terminal deoxynucleotidyl transferase (TdT), y se visualizan en el citómetro de flujo. Se trata de un método rápido que permite la determinación y cuantificación de células apoptóticas.
TINTES DE VIABILIDAD FIJABLES
Muchos de los reactivos utilizados para medir viabilidad celular, no son compatibles con la fijación, ya que durante este proceso se produce una permeabilización suficiente como para dejar pasar estos reactivos al interior celular, tanto en células muertas como en células vivas.
Actualmente se están desarrollando muchos reactivos que permiten la detección de células muertas en muestras fijadas, ya que cada vez existen más protocolos en los que se necesita permeabilizar y fijar las muestras (ej.: tinción intracelular).
Estos reactivos, a diferencia de los tintes de unión al ADN descritos anteriormente, se unen a grupos amino reaccionando con todas las proteínas celulares y, dado que son impermeables a las membranas celulares intactas, solo pueden unirse a las proteínas de la superficie de las células vivas. En el caso de las células muertas, la fluorescencia será mayor puesto que en el interior celular podrá unirse a un mayor número de proteínas.
Dado que estas moléculas pueden soportar las duras reacciones de entrecruzamiento asociadas con la fijación, se pueden usar dentro de los protocolos de fijación y permeabilización.
TIPS finales
¿Tu muestra está fijada o no?
Utiliza siempre tinte de viabilidad para mayor limpieza y sensibilidad del ensayo.
Elige un tinte de viabilidad compatible con tu panel.
Utiliza controles positivos (ej.: PFA) y negativos.
Titula las sondas de viabilidad: en exceso pueden resultar citotóxicas.
AUTORA
Ángela Marquina Rodríguez
Técnico Titulado Superior del Servicio de Citometría. Hospital Nacional de Parapléjicos. Toledo, España.
En la actualidad, la microscopía es una disciplina fundamental en las investigaciones biomédicas. Con ella se obtienen imágenes que posteriormente se analizan para obtener resultados. Dentro de las distintas formas de extraer información de las imágenes, en los últimos años ha habido grandes avances en el campo de la inteligencia artificial. En concreto, para el análisis de este tipo de imágenes se están empleando cada vez con más frecuencia algoritmos conocidos como redes neuronales artificiales (ANNs), que se engloban dentro del deep learning (DL). Como con toda nueva herramienta, es necesario tener precaución y validar correctamente los resultados obtenidos. Desde la comunidad de analistas de imagen científica se han propuesto distintos parámetros de medición que permitan realizar esta validación(1). En esta newsletter nos vamos a basar en esa publicación para describir estos parámetros y, con la ayuda de los Colab Notebooks del proyecto ZeroCostDL4Mic(2,3), los aplicamos para evaluar distintos modelos de ANNs entrenados en el SMAI, comprobando cómo se relacionan con la calidad de la predicción obtenida al aplicarlos tanto para la detección de objetos como para la mejora de imágenes.
Todos los parámetros de medición que aquí se describen permiten la comparación de la predicción generada por modelos de DL a imágenes etiquetadas o de ground-truth. Cuál elegir depende del tipo de tarea realizada por el modelo de DL. En algunos casos puede haber varios parámetros de medición disponibles que se pueden usar juntos.
Parámetros de medición de similitud de imágenes (eliminación de ruido, restauración y traducción de imagen a imagen)
Se pueden utilizar varios parámetros de medición para evaluar la similitud de dos imágenes:
El mapa RSE muestra la raíz cuadrada de la diferencia al cuadrado entre dos imágenes. En este caso, un RSE más pequeño es mejor. Una coincidencia perfecta entre la ground-truth y la predicción conducirá a un mapa RSE mostrando ceros en todas partes.
Donde P(i,j) es el valor de predicción en el píxel (i,j) y GT(i,j) es el valor del mismo píxel en la ground-truth. Habitualmente las imágenes se normalizan antes de la evaluación.
La raíz cuadrada del error cuadrático medio normalizado (normalized root mean squared error NRMSE) nos da la diferencia media entre todos los píxeles de las imágenes comparados entre ellos.
Donde N es el número total de píxeles. Habitualmente las imágenes se normalizan antes de la evaluación.
El coeficiente de correlación de Pearson (PCC) representa el grado de correlación lineal entre dos imágenes. Una correlación alta entre la predicción y la ground-truth produce un PCC cercano a 1.
El SSIM evalúa si dos imágenes contienen las mismas estructuras basándose en los conceptos de contraste, luminancia y contenido estructural. Es un parámetro de medición normalizado y un SSIM de 1 indica una similitud perfecta entre las dos imágenes. Los mapas de SSIM se generan calculando el valor SSIM en cada píxel pero también considerando los píxeles circundantes. El mSSIM es el valor de SSIM calculado en toda la imagen.
Pico de la relación señal-ruido (PSNR) estima las discrepancias entre dos imágenes con respecto al pico de la amplitud de la señal de la imagen de predicción. Suele calcularse en decibelios y cuanto mayor sea la puntuación, mejor será la concordancia.
En la siguiente figura podemos observar el resultado de usar estos parámetros en una imagen que ha sido tratada para reducir el ruido utilizando cuatro métodos distintos. En primer lugar, un desenfoque gaussiano de radio 1, un método tradicional de reducción de ruido (GAUSS). A continuación tenemos una imagen tratada con CARE(4), que entrena el modelo de ANN a partir de pares de imágenes con y sin ruido. Después tenemos el resultado de aplicar a la imagen «SOURCE» una ANN entrenada también con CARE, pero empleando pares de imágenes ruidosas para entrenar el modelo (Noise2Noise o N2N). Por último, usamos un modelo en el que en el entrenamiento partimos únicamente de imágenes ruidosas desemparejadas (Noise2Void(5) o N2V). Podéis ver más detalles de cómo se han generado estas imágenes y los modelos empleados en la newsletter de noviembre de 2020(6).
En la tabla de la figura podemos ver cómo con todos los métodos de reducción de ruido obtenemos valores mejores de los que se obtienen al comparar la imagen ruidosa (SOURCE) con la GT. Los mejores valores se obtienen con el empleo de CARE, seguidos por N2N. El desenfoque gaussiano produce unos valores solo un poco peores que N2N, pero son mejores que los obtenidos con nuestro modelo de N2V.
Parámetros de segmentación:
La segmentación de imágenes tiene como objetivo definir áreas de interés en una imagen en función de su identidad (elementos destacados frente al fondo es el más común). Una segmentación generalmente proporciona una imagen de máscara binaria donde los píxeles en el área segmentada tienen un valor de 1 (destacados) mientras que el resto de los píxeles tienen un valor de 0 (fondo).
El parámetro de medición IoU es un método que se puede utilizar para cuantificar la superposición entre dos máscaras binarias. Por lo tanto, cuando se utiliza IoU para evaluar el rendimiento de un algoritmo de segmentación comparando el resultado con las máscaras de la ground-truth, cuantomás próximo a 1, mejor habrá funcionado el algoritmo.
Donde ∪ representa la unión de dos imágenes binarias (número de píxeles destacados en cada una de las imágenes, ∩ representa la intersección de dos imágenes binarias (número de píxeles destacados en ambas imágenes a la vez) y P es la imagen predicha.
Parámetros para evaluación de la detección y clasificación de objetos (o instance segmentation):
La instance segmentation intenta detectar objetos en una imagen y distinguirlos tanto del fondo como del resto de objetos. De nuevo, hay varios parámetros que se pueden utilizar:
Es habitual calcular un valor de IoU entre la predicción y la ground-truth objeto a objeto. Esto permite identificar verdaderos positivos (NbTrue positive, normalmente con IoU>0,5), falsos positivos (NbFalse positive) y falsos negativos (NbFalse negative):
Donde NbGround-truth y NbPrediction son el número de objetos en la ground-truth y en la predicción, respectivamente.
La Precisión se define como el número de objetos segmentados correctamente dividido por el número total de objetos detectados. Permite valorar el coste de los falsos positivos. Cuanto más se acerque el resultado a 1, mejor será la predicción de la red neuronal.
El Recall calcula cuántos de los objetos positivos reales ha reconocido el modelo como verdaderos positivos. Se puede usar para evaluar el coste asociado a los falsos negativos. Cuanto más se acerque a 1, mejor.
El F1 combina los valores de Precisión y Recall en un único parámetro.
La mean matched score es la media de los IoU de los verdaderos positivos. La mean true score es el mismo valor, pero normalizado al número total de objetos en la ground-truth. La Panoptic Quality es el producto de F1 y la mean true score.
En la figura anterior vemos el resultado de aplicar a la imagen «Input» tres modelos de instance segmentation distintos entrenados con StarDist (para más detalles sobre StarDist podéis ver la newsletter(7) que publicamos anteriormente y el artículo original de Schmidt y col. 2018(8)). En la tabla podemos ver los valores de detección y segmentación para cada uno de los tres modelos. Observamos cómo el modelo C ofrece unos resultados bastante pobres tanto en la segmentación (con una IoU de 0,684), como en el reconocimiento de objetos (con gran número de falsos positivos y falsos negativos). En cambio, los modelos A y B producen unos resultados muy buenos y prácticamente idénticos, solo diferenciándose ligeramente en el valor de IoU (y, por consiguiente, en los valores que dependen de la IoU para su cálculo).
En resumen, en esta newsletter hemos visto cómo, con la ayuda de los de los Colab Notebooks del proyecto ZeroCostDL4Mic, se han evaluado distintos modelos de ANNs de una manera sencilla. Los parámetros de evaluación propuestos producen unos valores que permiten una fácil valoración de la validez de los modelos de ANNs , permitiendo la elección de los más apropiados. Además, los mapas de SSIM y RSE facilitan la detección de posibles artefactos en la reconstrucción o mejora de las imágenes.
Por último, quería resaltar que los Colab Notebooks de ZeroCostDL4Mic no solo se han usado para la evaluación, sino también para entrenar las distintas ANNs que hemos empleado hoy en los ejemplos. En una próxima newsletter os hablaré de este interesante proyecto que pone el uso de la IA para el análisis y mejora de las imágenes científicas al alcance de todo el mundo, ya que no requiere ni experiencia en programación ni un equipo informático especial y es completamente abierto y gratuito.
Referencias
Laine RF, Arganda-Carreras I, Henriques R, Jacquemet G. Avoiding a replication crisis in deep-learning-based bioimage analysis. Nat Methods. 2021 Oct;18(10):1136-1144. doi: 10.1038/s41592-021-01284-3. PMID: 34608322; PMCID: PMC7611896.
von Chamier L, Laine RF, Jukkala J, Spahn C, Krentzel D, Nehme E, Lerche M, Hernández-Pérez S, Mattila PK, Karinou E, Holden S, Solak AC, Krull A, Buchholz TO, Jones ML, Royer LA, Leterrier C, Shechtman Y, Jug F, Heilemann M, Jacquemet G, Henriques R. Democratising deep learning for microscopy with ZeroCostDL4Mic. Nat Commun. 2021 Apr 15;12(1):2276. doi: 10.1038/s41467-021-22518-0. PMID: 33859193; PMCID: PMC8050272
Weigert, M., Schmidt, U., Boothe, T. et al. Content-aware image restoration: pushing the limits of fluorescence microscopy. Nat Methods15, 1090–1097 (2018). https://doi.org/10.1038/s41592-018-0216-7
Jaakko Lehtinen, Jacob Munkberg, Jon Hasselgren, Samuli Laine, Tero Karras, Miika Aittala, Timo Aila. Noise2Noise: Learning Image Restoration without Clean Data. https://arxiv.org/abs/1803.04189