¿Has observado alguna alteración en tus células tras separarlas? ¿Cómo podemos conseguir sacar lo mejor de nuestras células separadas?
¿Qué es el SICS?
Sus siglas hacen referencia a Sorter Induced Cellular Stress y, por tanto, es el estrés que sufren las células separadas, a partir de una muestra homogénea, mediante un citómetro separador o cell sorting.
La mayoría de estos citómetros utilizan la separación electroestática para aislar varias poblaciones diferentes con elevada pureza y de forma simultánea.
Sin embargo, el procesamiento previo de la muestra (preparación, tinción, temperatura, etc.) junto con algunos parámetros de la separación (presión, descompresión/expansión, láser, cargas electroestáticas, etc.) someten a las células a numerosas fuerzas mecánicas que originan un estrés en las células aisladas.
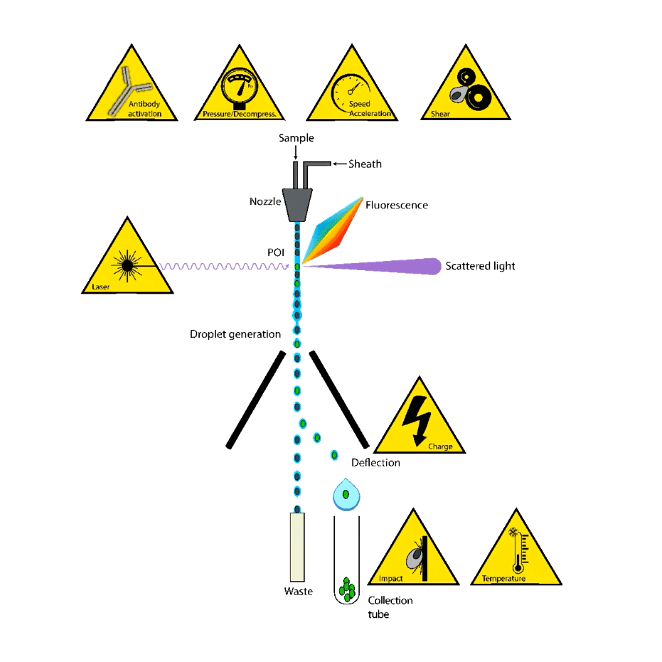
¿Qué puede provocar SICS?
- El proceso de preparación previo a la separación (disgregación de tejidos), puede volver a algunas células más susceptibles al SICS.
- La gran proporción de celulas muertas en la población inicial.
- Las condiciones dela separación tamaño del nozzle y presión, van de la mano en la mayoría de las ocasiones. Aunque también pueden cambiar dependiendo del instrumento y la configuración deseada.

Cuando las células son relativamente robustas, la presión puede ser un factor condicionante menor. Sin embargo, cuando son delicadas la presión debe ser la menor posible (Ej.: megacariocito: presión 20 psi y nozzle 100um).
Las células de gran tamaño pueden interrumpir la stream y originar aerosoles. Por ello, el diámetro del nozzle debe ser 3 veces mayor que el diámetro de la célula. Por tanto, es necesario conocer el tamaño de las células que queremos aislar.
- El uso de tampones incompatibles.
El tampón en el que se encuentra la muestra (o sample tampón) puede ser HBSS, medio de cultivo (sin rojo fenol, puesto que puede dar autofluorescencia), PBS (sin Ca2+/Mg2+) o cualquier otro tampón siempre y cuando contenga los siguientes aditivos:
- Fuente de proteínas- FBS y/o BSA.
- HEPES para regular el pH.
- EDTA para evitar los agregados celulares.
- DNase para degradar los resto de DNA de las células muertas (cuando haya mucha muerte celular en el procesamiento).
El uso de medio de cultivo como tampón de recogida (o collection tampón ) NO está recomendado, ya que suelen tener en su formulación cloruro de calcio que podría interferir con los fosfatos que componen el fluido envolvente y dar lugar a cristales de fosfato que pueden dañar las células separadas que se encuentran en el tubo de recogida.

- Duración dela separación. Las células están sometidas a presión durante largos periodos de tiempo.
¿Cómo podemos identificar que nuestras celulas están sufriendo SICS?
- La morfología de las células es diferente antes y después de la separación o sorting.
- Retraso del crecimiento celular después de la separación (post sort). Las células separadas no crecen tan rápido como las que no han sido aisladas.
- Alteraciones en las funciones de las celulas separadas, pueden retrasarse, disminuir o incluso desaparecer.
- Se producen cambios en el fenotipo transitoria o incluso, permanentemente.
¿Cómo podemos minimizar SICS?
- Tras la separación las células forman agregados celulares, debris, mueren -> Esto quizás se deba a que las células no estén bien desde el primer momento. Usa sondas de viabilidad con el fin de monitorizar la muerte celular inicial.
- Las células mueren -> Intenta:
- separar en frío
- incrementa la concentración de suero en el tampón de separación
- usa tampones similares
- usa un nozzle de mayor tamaño.
- Disminuye el tiempo de separación aumentando la velocidad (nozzle más pequeño=mayor presión= menos tiempo de separación=menor volumen recogido y menor dilución del tampón de recogida)
- La pureza es baja después de cultivar las células separadas -> Las células seleccionadas pueden no crecer tan rápido como las células contaminantes que pueden estar presentes tras la separación.
- ¿Todavía tienes problemas? -> Cambia la tinción para que la población de interés sea “negativa”.

Resumen…
- Comienza con una buena preparación de la muestra.
- Sigue las buenas prácticas.
- Conoce tus células (observa morfología y tamaño al microscopio antes y después de la separación).
- Comprende qué requieren las células.
Fuente: “Cell Sorting for Function and Viability”. Seminario impartido por CYTO University (Purdue) International Society for Advancement of Cytometry (ISAC) online. http://cytou.org/store/seminar/seminar.php?seminar=125786

