En el artículo del 20 de abril os hablamos de un plugin de FIJI (Trainable WEKA segmentation)que utiliza el machine learning para ayudarnos a segmentar las imágenes clasificando los píxeles en distintas categorías de interés. Hoy os queremos hablar de otra herramienta (StarDist) que utiliza redes neuronales convolucionales, dentro del campo del deep learning (un método más avanzado de machine learning), para ayudarnos a identificar objetos individuales en nuestras imágenes.
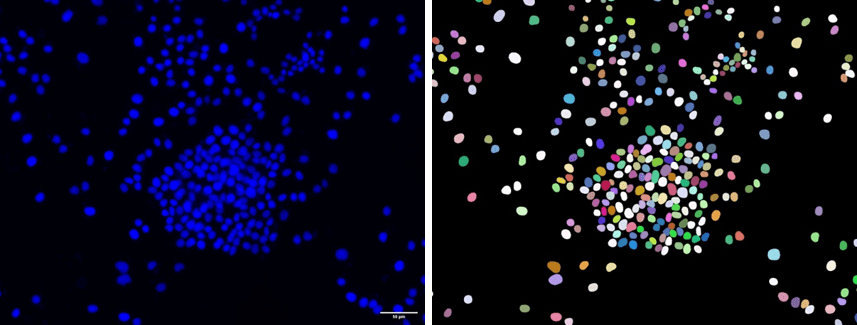
Ejemplo de identificación de núcleos en los que a todos los píxeles de cada núcleo individual se les asigna un valor único, obteniendo lo que se conoce como imagen etiquetada o labeled image (derecha).
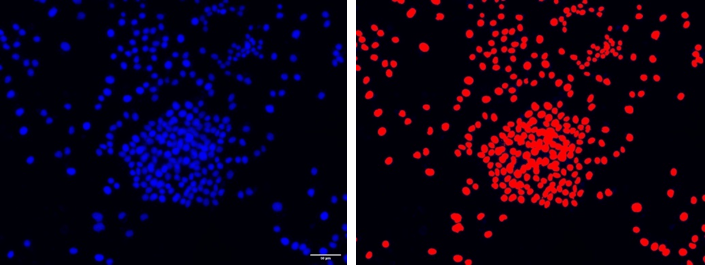
Si bien en el ejemplo anterior bastaría con aplicar un umbral de intensidad automático para separar los núcleos, con frecuencia es mucho más complicado hacer la segmentación (véase la imagen siguiente):
Los núcleos en esta muestra están demasiado juntos como para poder separarlos usando un umbral de intensidad (derecha).
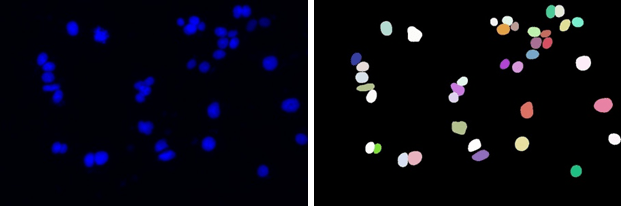
En casos como este, podemos usar StarDist para entrenar una red neuronal que aprenda a distinguir los objetos individuales. Básicamente, lo que tenemos que hacer es preparar una serie de imágenes de entrenamiento junto con su solución en forma de imágenes etiquetadas, que serían lo que se conoce como ground truth.
Ejemplo de imagen de entrenamiento (izquierda) y su ground truth (derecha).
El etiquetado de las imágenes de entrenamiento se hace normalmente a mano (si se pudiera hacer automáticamente no sería necesario usar StarDist) y es la parte del proceso que puede llevar más tiempo. El número de imágenes necesarias para el entrenamiento va a depender de la variabilidad de las imágenes a analizar pero puede funcionar con tan pocas como 5-10 con 10-20 objetos anotados en cada una.
Una vez entrenado el modelo, conviene ponerlo a prueba en un grupo de imágenes distintas a las que se han usado para entrenar pero de las que también tenemos su ground truth. De este modo podemos evaluar su precisión. Si no estamos satisfechos con los resultados, tendremos que incrementar el número de imágenes y/o el número de ciclos de entrenamiento hasta que obtengamos una precisión aceptable.
Con el modelo entrenado, podemos usar el plugin de StarDist para FIJI para analizar nuestras imágenes. De hecho, este plugin ya viene con una serie de modelos preentrenados que se pueden usar.
A continuación podéis ver el resultado del análisis de una imagen usando el modelo preentrenado:
Aunque el resultado parece bastante bueno a primera vista, si os fijáis os daréis cuenta de que existen bastantes falsos positivos (algunos están señalados con flechas amarillas). Aun así, si tenemos en cuenta que las imágenes usadas para entrenar el modelo incluido en el paquete de StarDist se han adquirido con condiciones y equipos distintos de los que se han usado para la adquisición de esta imagen, nos podemos hacer una idea del potencial que tiene esta técnica de análisis. Como podéis ver en la siguiente figura, cuando entrenamos el modelo usando imágenes adquiridas en las mismas condiciones y con el mismo equipo, el resultado mejora hasta ser casi perfecto.
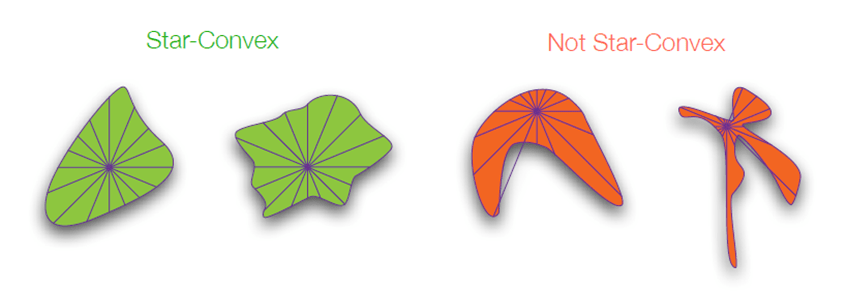
StarDist está diseñado para detectar objetos con una forma que se describe como star-convex. Se podrían definir como objetos que tienen algún punto dentro de ellos desde el que se pueden ver todos sus límites. Esto lo hace perfecto para detectar objetos de una forma más o menos redondeada, como los núcleos celulares o células sin protrusiones citoplasmáticas.
Si queréis saber más sobre esta herramienta y si sería una opción para el análisis de vuestras muestras, no dudéis en poneros en contacto con nosotros.
También podéis obtener más información en la página de StarDist (https://github.com/mpicbg-csbd/stardist) – donde tenéis acceso a una descripción de los fundamentos teóricos de su funcionamiento y el acceso a los artículos originales (de 2018 y 2020) – y en el muy interesante webinar de NEUBIAS @Home donde se entra más en detalle a hablar los conceptos y aplicaciones prácticas de la herramienta (https://www.youtube.com/watch?v=Amn_eHRGX5M).
Por último, solamente indicaros que existen otros modelos de deep learning que no están restringidos al reconocimiento de objetos con formas star-convex. Si estáis interesados en conocer más sobre ellos, hacédnoslo saber y quizá podamos presentarlos en un futuro artículo de la newsletter.
Agradecemos al Grupo de Química Neuro-Regenerativa del Dr. Ernesto Doncel que nos haya permitido usar las imágenes de sus muestras para esta newsletter.
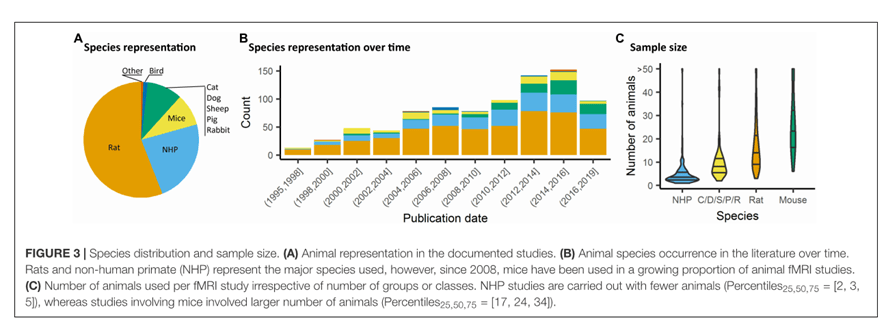
En este segundo capítulo del Servicio de Resonancia Magnética de Investigación queremos hablaros en una pequeña introducción de la imagen funcional por Resonancia Magnética, la cual permite estudiar la funcionalidad, conectividad y actividad cerebral.
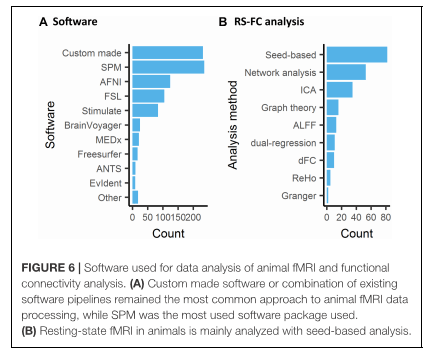
A dia de hoy, hay una gran variedad de estudios de fMRI en los que se varía el tipo de anestesia, las secuencias de adquisición, los paradigmas empleados; todo para definir/o unificar un buen diseño experimental, ya que en imagen animal siguen surgiendo controversias tanto en el método de análisis como el tratamiento de las imágenes. Son muchos los artículos publicados y solo 868 los que se han considerado relevantes para escribir la revisión en la que se basa este documento. La revisión se ha hecho en función del tipo de estudio: Resting State, neuromodulación opto/quimiogenética provocada por fármacos, estimulación profunda, o estimulaciones provocadas (por bloques o por eventos con estimulación sensorial, o gaseosa, etc.). Se ha escrito también sobre la especie animal, número de animales, la preparación del animal: despierto, anestesiado con y sin ventilación, el nivel de anestesia durante la adquisición, el campo magnético, la secuencia de fMRI y el contraste, y los programas de pre-procesamiento.
A continuación se detalla alguno de los puntos más relevantes de la revisión.
El diseño experimental
Imagen fMRI con estímulos provocados
Se pueden emplear estímulos externos (p.ejemplo: estimulación eléctrica de la pata delantera) o también internamente (estimulación profunda cerebral (DBS) y optogénetica). Los estímulos se pueden aplicar en bloques o en un diseño de eventos relacionados. En el primero se alternan condiciones de estimulación +/-, mientras que en el segundo se aplica un pequeño estímulo en varios intervalos.
En la estimulación por bloques se estudia la frecuencia relacionada con las respuestas producidas y en el de estímulos relacionados se estudia la duración del tiempo/respuesta y así poder valorar la conectividad funcional independiente de la frecuencia.
Las altas frecuencias aumentarán la entrada de estímulos por unidad de tiempo, aumentando la señal y la habilidad de detectar respuestas provocadas. Esta estimulación eléctrica u óptica, puede provocar daños en el tejido (calentamiento), artefactos y efectos no específicos. También puede cambiar la fisiología básica y por tanto alterar la respuesta fMRI.
Mediante esta técnica, los datos que se recogen son sin estimulación. Se emplea para investigar la sincronización de las señales que varían espontáneamente en una combinación anatómica y funcional en unas áreas de cerebro determinados.
El empleo de esta técnica se ha incrementado a lo largo de esta última década. En los estudios recientes, sugieren que los componentes de la red cerebral muestran unas propiedades no estacionarias, el tiempo de repetición tiene que ser corto (1seg) para recoger las fluctuaciones y detectar estos cambios. El tiempo de adquisición total deberá ser lo suficientemente largo para adquirir un mínimo de 300 imágenes.
También hay que tener en cuenta, que si se aplica una modulación/estimulación hay que añadir un tiempo durante los periodos de transición y el resting state para permitir una conectividad estable, y por tanto, un tiempo de reposo que permita que cada manipulación pueda ser agrupada separadamente para tener en cuenta las potenciales neuroadaptaciones. Hay que trabajar con cuidado con la fisiología y la anestesia para estar seguros de que se consigue la máxima detección de la señal.
Optogenética
Permite una estimulación en una población específica celular y/o anatómica pero requiere una metodología muy rigurosa y controles adicionales. Las opsinas son las proteínas que actúan como canales/bombas que son activadas por la luz. Hay varias opsinas que pueden ser elegidas para excitar las células ópticamente, entre ellas la más comúnmente utilizada es la ChR2 que se activa con luz roja y variantes ultra rápidas capaces de aumentar la frecuencia a 200Hz. También hay opsinas que producen una inhibición celular, pero su aplicación a la fMRI está limitada porque requieren largos periodos de iluminación que producen artefactos relacionados con el calor, y donde los animales anestesiados o sedados dan bajas señales de actividad. La inyección de virus o la expresión de genes pueden cambiar potencialmente la función cerebral y la luz puede inducir calor y con ellos artefactos en al señal de IRM, daño tisular y efectos no específicos que son críticos para caracterizar la expresión de opsinas.
Quimiogenética
Inicialmente llamada farmacogénetica, se emplean ligandos farmacológicamente inertes para producir una estimulación en unos receptores específicos sobre una población neuronal específica. Los primeros intentos para combinar esta aproximación con la fMRI han involucrado la expresión regional de receptores de proteínas G acopladas farmacologícamente activadas. Esta metodología al igual que en la optogenética, requiere una metodología muy rigurosa para controlar los efectos no deseados.
fMRI con fármacos
La modulación del cerebro por fármacos durante la fMRI es ampliamente utilizada para estudiar los efectos globales de los compuestos en el sistema neurotransmisor diana. No requiere cirugía y es apta para identificar los cambios globales o regionales de la función asociados a las nuevas terapias que actúan sobre las enfermedades cerebrales producidas a nivel de neurotransmisores o para mapear el efecto de los neuromodulares administrados. Sin embargo, esta metodología no es la más apropiada para estudios dinámicos y repetitivos ya que los efectos son dependientes de la difusión y de la cinética del receptor que pueden llevar a una desensibilización y baja regulación del receptor. Hay que tener en cuenta los efectos dosis-respuesta y la farmacocinética del compuesto en el diseño experimental. Muchos agentes tienen efectos sistémicos que pueden afectar a la fisiología del animal y a la señal BOLD. Algunos fármacos tienen efectos directos en el endotelio vascular del cerebro que podría alterar las propiedades de la respuesta hemodinámica.
Es muy importante llevar un control y una monitorización de la fisiología del animal y usar una dosis apropiada para controlar los efectos secundarios. El aumento del flujo y volumen sanguíneo y un incremento en la presión sanguínea producido en las infusiones puede alterar la señal de MRI.
Factores a tener en cuenta:
Tipo de especie, el tamaño de muestra, sexo del animal:
En la revisión llevada a cabo los estudios que se han realizado han sido sobre muchas especies, siendo la rata la más utilizada, concretamente la especie Sprague Dawley
La descripción de cada arquitectura funcional en cada especie se ha basado en una variedad de análisis, adquisiciones y protocolos con/sin anestesia. Esta falta de estandarización interespecie es debida al tamaño del cerebro, a las diferentes respuestas frente a la anestesia, y a las organizaciones anatómicas observadas entre los mamíferos.
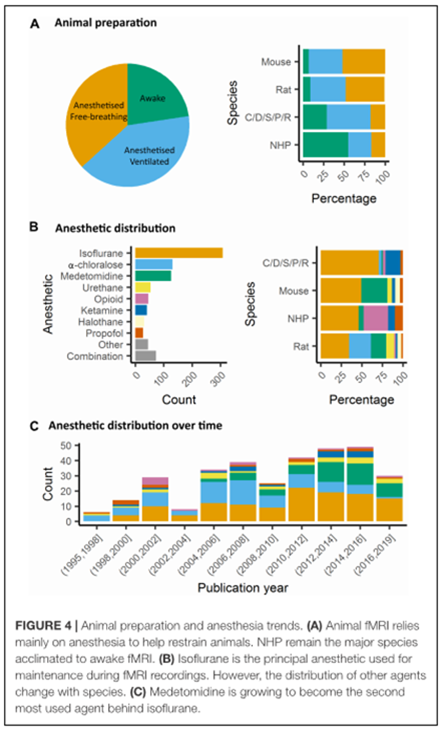
Preparación del animal y anestesia
Influencia del movimiento y el estrés.
En fMRI la información obtenida es debida a los cambios temporales en los parámetros hemodinámicos (oxigenación de la sangre: contraste BOLD, volumen sanguíneo cerebral CBV, o flujo sanguíneo cerebral (CBF). Las señales fMRI explicarían la activación neuronal a través de la evaluación de la respuesta hemodinámica, por ejemplo la adaptibilidad de los capilares locales para entregar oxígeno a las neuronas activas a una velocidad mayor que las neuronas inactivas. La señal BOLD es el parámetro más estudiado en fMRI, es dependiente de los niveles relativos de oxihemoglobina y deoxihemoglobina, modulados a su vez por los volúmenes y el flujo de sangre a nivel local.
Además las adquisiciones de fMRI son muy sensibles al movimiento del sujeto, especialmente en los límites del tejido. En la mayoría de los estudios, los animales son anestesiados para evitar el movimiento, se sujetan también a través de barras en los oídos y en los dientes. Pero también hay estudios con el animal despierto, empleando dispositivos que les retenga en un espacio más limitado utilizando barras que sujeten la cabeza y que no les cause ningún tipo de daño ni malestar. Pero por mucho que se fije el animal siempre hay un movimiento que es el de la respiración; para ello, se puede recurrir a la ventilación mecánica del animal, de esta manera se reduce de manera considerable el artefacto de movimiento durante la adquisición de la señal. Otra fuente de ruido y mala interpretación de la señal, es el producido por el latido cardiaco que induce pequeñas fluctuaciones en la señal BOLD a bajas frecuencias.
En algunos casos, la respuesta cardiaca puede enmascarar la respuesta neuronal, especialmente la respuesta a un estimulo de estrés.
Impacto de la anestesia en la fisiología animal
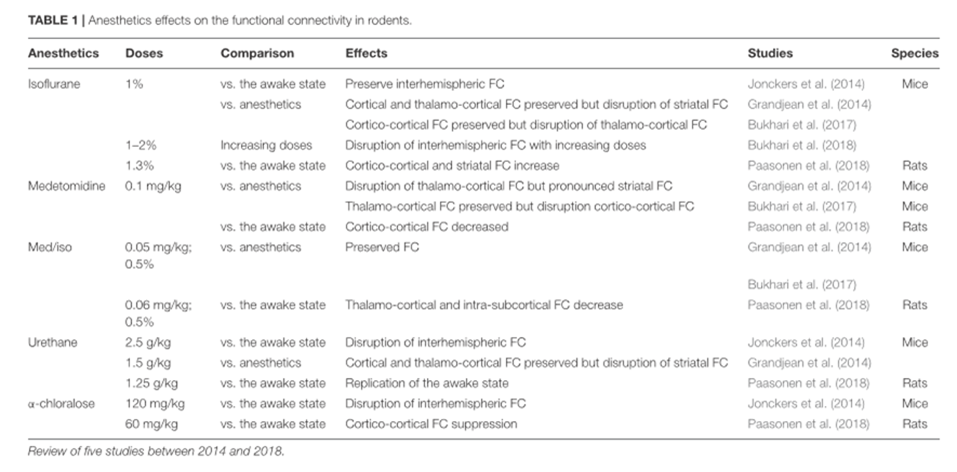
La señal BOLD está modulada por el latido cardiaco, la concentración de CO2 arterial y la temperatura. Existe una gran variedad de anestésicos que modulan diferentes dianas en el cerebro y que actúan sobre los receptores periféricos causando una regulación cardiaca y respiratoria. Por tanto, producirán distintos efectos sobre la señal BOLD y sobre otros parámetros hemodinámicos.
Por ejemplo: en una rata anestesiada con ventilación: donde paCO2, paO2 y pH se mantienen constantes, se víó una reducción del contraste T2* entre las venas y el parénquima cuando la rata estaba anestesiada con Isofluorano al 2% en lugar de medetomidina o ketamina/xylacina. Con el isofluorano se incrementa el CBF y la vasodilatación.
El mantener al animal ventilado hace que se evite la hipercapnia (aumento de paCO2) que tiene un efecto sobre la reproducibilidad del estudio fMRI . La hipercapnia también produce una vasodilatación y un incremento en el CBF. La variación del CBF puede explicar también una disminución especifica de la respuesta BOLD en la actividad neuronal inducida por un estímulo.
En la revisión, se han estudiado los efectos de diferentes anestésicos sobre la oxigenación sanguínea en diferentes regiones cerebrales. El impacto de la anestesia sobre otros parámetros fisiológicos, tales como la temperatura corporal y actividad cardiovascular periférica pueden modular la calidad de la conectividad funcional. El mantenimiento de estos parámetros pueden ser controlados y monitorizados. Otro de los parámetros que deben de ser controlados es la presión sanguínea.
Los anestésicos empleados para los estudios con animales se han clasificado en función de su diana: los que actúan sobre los receptores GABAa, NMDA, canales de K+ .
Los más empleados son los que actúan a través de los receptores GABAa: son canales de Cl- que hiperpolarizan las neuronas, haciendo que sean menos excitables e inhibiendo los potenciales de acción. Los anestésicos más empleados son el Isofluorano, Propofol y barbitúricos. Cada fármaco actúa de distinta manera
El isofluorano y Sevofluorano tienen actividad metobólica opuesta en el flujo sanguíneo cerebral y distinto metabolismo de la glucosa en varias regiones del cerebro La alfa-cloralosa también es utilizada porque produce una buena respuesta metabólica y hemodinámica.
Otro de los antagonistas de los receptores de NMDA empleados es la ketamina que bloquea la actividad sináptica excitatoria. Produce un aumento de la actividad cerebral regional y un aumento del volumen sanguíneo cerebral. En roedores los más utilizados son el Isofluorano y la medetomidina
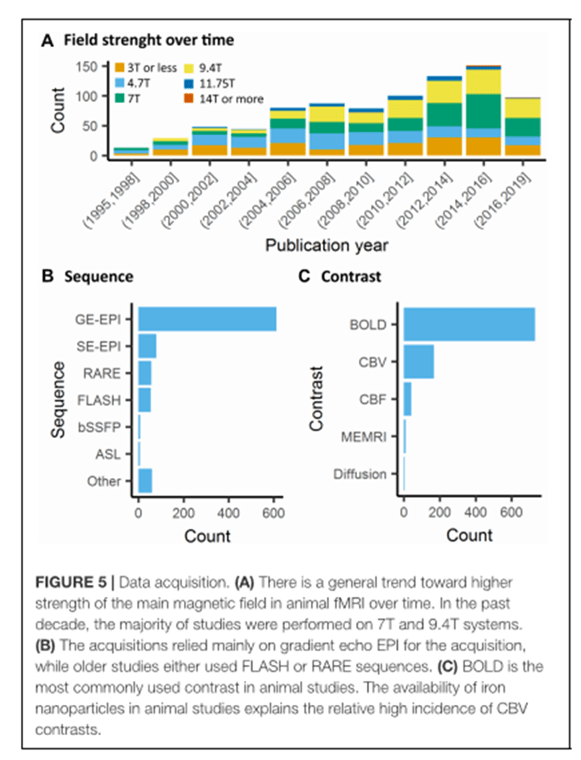
Adquisición de los datos
La actividad neuronal induce una vasodilatación en los capilares y arteriolas vecinas que se puede propagar hacia las arterias y venas. El aumento del CBF y CBV y la oxigenación de la sangre es en lo que se basa el principio de la fMRI. El método más común es el contraste BOLD: en el que se estudian las propiedades paramagnéticas de la deoxihemoglobina que produce un cambio en la susceptibilidad magnética dentro de los vasos sanguíneos y tejido vecino que es detectado por secuencias de T2 y T2*.
La [deoxiHb] aumenta desde las arterias (<5%) hacia las venas (40%) en el árbol vascular debido a la extracción del oxígeno de los capilares haciendo que la imagen BOLD sea particularmente sensible en los capilares, vénulas y venas. En el tejido cerebral sano, la actividad neuronal induce un aumento del CBF que produce un aumento de la entrega del oxigeno que excede sobre la disminución del oxigeno en los capilares. Como resultado, la [desoxiHb ] en los capilares y venas decrece, dando una señal BOLD positiva detectada en las imágenes de T2 y T2*.
Las secuencias más empleadas en los estudios de fMRI en roedores son las de eco de gradiente pero es muy sensible a los artefactos de susceptibilidad, más en las cavidades cercanas a los oídos y bulbo olfatorio, particularmente cuando se utilizan tiempos de eco largos en campos magnéticos altos. Esta secuencia también es muy sensible a las venas grandes que hace que la secuencia no sea espacialmente precisa en los acoplamientos neurovaculares a nivel de los capilares. Para obtener una actividad neuronal espacial específica se usa la secuencia de eco de espin que es sensible a los vasos sanguíneos pequeños.
Aunque el contraste BOLD es el más empleado para hacer fMRI, otro de los métodos que sirve para calcular el CBV y/o el CBF es el empleo de agentes de contraste exógeno con partículas de óxido de hierro. Estas nanopartículas tienen una relajatividad r2 y r2* muy alta, no atraviesan la barrera hematoencefálica cuando está intacta y tienen un tiempo de circulación en sangre de horas. La administración intravenosa de éstas produce fenómenos de susceptiblidad magnética dentro de la vasculatura y en los tejidos de alrededor, que a una dosis alta tienen muchos más efectos que la deoxihemoglobina. Como resultado, la señal T2* intravascular apenas se detecta, mientras que la señal T2* extravascular se hace más sensible a los cambios en el CBV. Un incremento en el CBV, inducido por una actividad neuronal, aumenta la susceptibilidad magnética dentro del voxel, dando un contraste T2* negativo-CBV no dependiente. El contraste CBV es independiente del campo magnético y produce una bajada de la intensidad de la señal con respecto a la línea base cuando se inyecta una nanopartícula de óxido de hierro.
Aunque la mayoría de las técnicas de fMRI en animales están basados en la respuesta hemodinámica a la actividad neuronal, existen otros métodos más específicos para estudiar esta respuesta neuronal: como la imagen de difusión-fMRI y la que emplea Manganeso, pero estas técnicas lo dificultan por la limitada o no uniforme sensibilidad, baja resolución espacial y por dudas en los mecanismos subyacentes.
ANALISIS DE DATOS
Pre-procesamiento
Incluye una serie de pasos para corregir los artefactos y normalizar los datos: Corrección de movimiento, Filtro temporal, Co-registro a un patrón de referencia. Son muchos las herramientas que se utilizan para este primer paso, pero hasta la fecha no existe un método universal para todos los estudios que se realizan en diferentes centros, por lo que los métodos empleados hacen difícil compararlos entre ellos.
Selección de patrón y del atlas
El registro de los resultados de Fmri a una referencia común es uno de los pasos necesarios en el preprocesamiento y permite que el análisis sea imparcial a nivel de voxel.
En los estudios de animales se ha comprobado que este paso en la mayoría de ellos no se hace, a pesar de que este paso proporciona un registro óptimo para el contraste de la imagen, resolución y orientación, esto añade desafíos extra para compararlos entre estudios.
Analisis resting state
Para los estudios de Fmri con: estimulos provocados, farmacológicos, DBS y optognética/quimicogenetica, la mayoría de ellos son analizados con estadística a nivel de voxel donde las series de tiempo en cada voxel son tratados como modelos independientes, normalmente se ajusta a un modelo de respuesta hemodinámica frente a un paradigma de estimulo/inyección. A menudo se complemente con un análisis de ROIs.
Como en en resting-state no se emplea un paradigma, se tienen que emplear modelos intrínsecos para estudiar la conectividad o medidas asociadas. Hay varios métodos de análisis que han sido desarrollados; primero en humanos pero que se pueden aplicar a los estudios con animales. Los análisis se basarían en hipótesis (crecimiento en semilla) hasta trabajar con los datos (análisis de componentes principales) y podrían ser aplicados a nivel de la red neuronal hasta un ROI en particular. Algunos métodos describen la relación entre áreas, otros se basan en las características de las variaciones a baja frecuencia de la señal BOLD en una sola región.
Bibliografía:
Animal Functional Magnetic Resonance Imaging: Trends and Path Toward Standardization Neuroinform., 22 January 2020: REVIEW ARTICLE
¿Qué es el estrés en un animal de laboratorio? ¿Y el sufrimiento? ¿Y el dolor?
Antes de saber las respuestas a estas preguntas tenemos que saber cuándo un animal se encuentra en óptimas condiciones físicas y mentales. En este caso, se encuentra en un estado de bienestar cuando hay una armonía entre el animal y el ambiente que le rodea. Esta armonía puede ser interrumpida debido a factores externos produciendo un estrés, que según las Directrices para el Reconocimiento y evaluación del dolor en Animales es un estado donde el animal tiene que dedicar un esfuerzo sustancial o recursos para la respuesta adaptativa a desafíos que emanan de la situación ambiental [1], es decir, la alteración del estado en el que el animal está en armonía consigo mismo y el entorno en unas condiciones extremas.
Estás alteraciones del bienestar del animal pueden producir diversas situaciones en las que dicho animal se puede adaptar a ese cambio que ha sufrido su entorno conllevando un coste ante una respuesta intensa o duradera de estrés, o puede no adaptarse en cuyo caso el resultado será muerte, lesiones o enfermedades por parte del animal.
En ambos casos el animal tendrá un sufrimiento que se producirá cuando el umbral de la tolerancia al dolor se vea superado como consecuencia a ese estrés o dolor, siendo por lo tanto, un estado mental,un malestar sensorial o experiencia emocional asociada con un daño actual o potencial y descrito en términos de perjuicio o daño.
También hay que saber diferenciar cuando un animal siente dolor, una experiencia sensorial y emocional desagradable asociada con un daño real o potencial, o sufrimiento; y cuando no. Por ejemplo, los cerdos reaccionan de forma muy estridente cuando se procede a una inmovilización. Esta reacción no es porque se les esté ocasionando un daño, sino porque es una especie que el contacto corporal es muy común y necesitan producir una señal sonora muy estridente para evitar que otros animales los aplasten debido a su tamaño.
¿Qué situaciones pueden causar estrés, dolor o sufrimiento?
Las situaciones que pueden causar dolor o sufrimiento a un animal entre otras, pueden ser:
Ejercicio forzado
Procedimientos invasivos y manipulación
Radiación, inhalación o ingesta de tóxicos
Exposición a variaciones de temperaturas extremas
Enfermedades propias o provocadas
Interrupción del ritmo circadiano
Brusquedad del personal u operario
Traumatismos
Abusos entre animales de la misma jaula, hacinamiento, sufrimiento o muerte de otros animales
El problema radica en que un animal no puede expresar verbalmente ese dolor o sufrimiento para que los operarios que los manipulan puedan poner remedio, por lo que dichos operarios deberán estar lo suficientemente entrenados para poder ser capaces de distinguir cuando un animal se encuentra saludable y cuando no, y detectar los síntomas que demuestran que el animal está sufriendo.
¿Cuáles son los síntomas de dolor y/o sufrimiento?
Estos síntomas según la Guía para el reconocimiento del dolor, estrés y disconfort presenta un listado de signos claves como aquellos:
Relacionados con la nutrición: deshidratación, heces irregulares…
Relacionadas con la actividad motora/refleja: parálisis
Relacionadas con el ritmo de actividad: aletargamiento
Relacionadas con el comportamiento general: ausencia de motivación
Más específicamente los signos fisiológicos clave de los ratones frente a estímulos dolorosos o estresantes descritos en Ciencia y tecnología del animal del laboratorio, son:
Oculares: Respuesta pupilar, los párpados pueden estar cerrados o semicerrados. En estados más avanzados podemos encontrar ojos hundidos o secreciones.
Respiratorio: El ritmo aumenta y se pueden producir secreciones nasales.
Aspecto: Piloerección, pérdida de peso y apariencia, deshidratación, lordosis, abdomen hundido por falta de alimentación y escaso o nulo acicalamiento.
Defecación/orina: ambos están afectadas por el estrés aumentando o disminuyendo.
Vibrisas (bigotes): aumentan el movimiento
Comportamiento: cada vez más tímidos y aprensivos, con tendencias agresivas. Cuando la condición empeora se muestran inactivos, sin respuesta y aislados. Eventualmente no muestran reacciones a estímulos.
Actividad anormal: aislamiento, mordeduras, lucha o enfrentamientos, deja de comer y beber y aumentan las heridas.
Postura: Gradualmente encorvado, dormitando.
Locomoción: Si hay dolor en patas, estas se verán afectadas provocando desplazamientos inseguros, dificultad para andar, movimientos circulares cuando el equilibrio se ve afectado.
Vocalización: agresiva al inicio del dolor y luego decrece.
General: Hipotermia y deterioro del estado físico.
En resumen, los signos a destacar para esta especie se establecen en reflejo de alejamiento, piloerección, encorvamiento, mordiscos, ojos y abdomen hundidos, pérdida de peso y deshidratación principalmente.
En el caso de las ratas, los signos fisiológicos que destacan frente a estímulos de dolor son:
Oculares: Párpados semicerrados. Ojos con apariencia hundida con secreciones de hematoporfirina.
Respiratorio: Aumento asociada con estornudos y secreciones nasales.
Aspecto: Piloerección, aspecto en el pelo desaliñado con aparición de calvas. Perdida del tono muscular, deshidratación y pérdida de peso.
Defecación/orina: Aumenta o disminuye según el estímulo o ingesta de alimentos y agua.
Conducta: inicialmente la respuesta suele ser agresiva con tendencia a morder.
Actividad anormal: Aumenta la somnolencia, disminución de la ingesta de alimentos y agua, dejan de explorar, en ocasiones se producen automutilaciones en zonas afectadas al final del tratamiento o manipulación.
Postura: Si el dolor es abdominal gradualmente asumirá una posición encorvada con la cabeza en el abdomen.
Locomoción: Si el dolor está localizado en las extremidades habrá cojera con movimientos cuidadosos, tensos, lentos o circulares dependiendo de la localización del dolor.
Vocalización: agresiva en la manipulación reduciéndose gradualmente.
General: Hipotermia, aspecto pálido que pueda indicar pérdida de sangre o anemia.
En resumen, los signos a destacar para esta especie son vocalización, lucha, acicalamiento, pérdida de peso, piloerección y lordosis junto con movimientos defensivos.
¿Qué se puede hacer para remediar el dolor y/o sufrimiento del animal de laboratorio?
Teniendo en cuenta todolo expuesto anteriormente, se deben establecer protocolos para que en el momento que se presente algún síntoma se proceda a la administración de analgésicos compatibles con las investigaciones o estudios que se estén realizando en ese momento con el animal.
En algunos tipos de experimentos se estudia la posibilidad de tratar al animal 24 o 48 horas previa y/o posteriormente a la cirugía para reducir estos síntomas en la medida de lo posible.
Según el artículo Refinamiento en el uso del animal: evaluación y alivio del dolor y sufrimiento, la administración repetida de estos analgésicos puede ser:
Inoculada: en cuyo caso se precisará de personal cualificado, además de inyectar la sustancia en diversas dosis diarias evitando en la medida de lo posible más molestias al animal puesto que la manipulación, inmovilización, etc… ya ocasionarán un estrés aumentando la situación de dolor que ya padece por lo que la respuesta del animal será más agresiva de lo normal.
Ingerida: a través de gelatinas especiales que contengan el analgésico.
Bebidas: en cuyo caso el analgésico estará diluido en el agua de bebida en la proporción recomendada.
La medicación usada para la reducción del dolor en roedores son analgésico cuyo principio activo son opiáceos o buprenorfina.
Un punto muy importante a tener en cuenta, es que el grado de dolor o sufrimiento que pueda causar cualquier tipo de procedimiento estará en gran medida influenciado por la experiencia del personal que esté involucrado, por lo que es muy importante que dicho personal sea cuidadoso y experimentado en este tipo de trabajo.
Hay que tener en cuenta que una mala manipulación o una mala gestión de las técnicas que se están realizando para llevar a cabo un procedimiento, podría ocasionar más lesiones o incluso la muerte del animal, resultando en una pérdida de muestras para la investigación y por lo tanto posibles resultados.
Referencias bibliográficas:
FELASA Working Group on Pain and Distress (1994) Pain and distress in laboratory rodent and lagomorphs.LaboratoryAnimals 28: 97-112
SECAL. Refinamiento en el uso animal: evaluación y alivio del dolor y sufrimiento. Laboratory Animals 28: 222-231
SECAL. Ciencia y tecnología del animal de Laboratorio (2008) Capítulo 13, 411-431
Las vesículas extracelulares (EVs) son pequeñas partículas rodeadas por una bicapa lipídica que son secretadas por todo tipo de células, y que constituyen un mecanismo de comunicación intercelular muy importante. Todas las células que se encuentran creciendo activamente o en una situación de estrés, van a secretar EVs al medio externo.
A lo largo de los años las EVs han recibido multitud de nombres, pero actualmente se recomienda la división en exosomas y microvesículas o ectosomas, atendiendo a su rango de tamaños y a su ruta de biogénesis.
Los exosomas son vesículas de entre 40-100 nm de diámetro secretadas a partir de cuerpos multi-vesiculares (MVB: “Multi-Vesicular Bodies”) generados en el interior celular a partir de endosomas, mientras que las microvesículas o ectosomas tienen tamaños entre 100 nm y 1 µm de diámetro y se producen por evaginación directa de la membrana plasmática.
Ambos tipos de EVs comparten distintas características, lo que a veces hace difícil su distinción o purificación por separado. Lo que sí está claro es que la composición de las EVs no es aleatoria y va a reflejar el estado de la célula que las produce, existiendo diversas moléculas y grupos de moléculas que se encuentran enriquecidas en las mismas. El contenido de las EVs se denomina CARGO y va a ser el responsable de los efectos que tengan esas EVs sobre otras células o tejidos, de ahí que una de las principales finalidades del estudio de las EVs sea el conocimiento de su cargo específico.
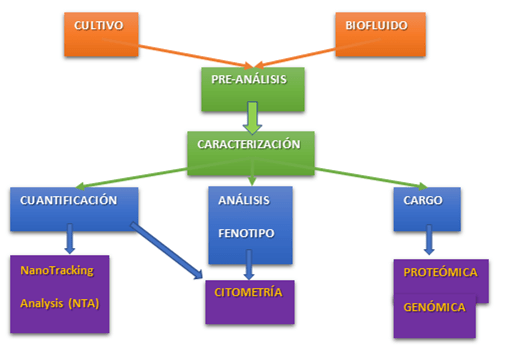
Por tanto, el estudio de las EVs va a incluir tanto la caracterización de las mismas en cuanto a nº de partículas, fenotipo, presencia en condiciones normales o patológicas, etc, así como la caracterización de su cargo específico mediante identificación de RNAs y/o estudios proteómicos.
Con el fin de realizar estudios lo más estandarizados posible y con unos controles de calidad adecuados que aseguren una correcta identificación de EVs, la Sociedad Internacional de Vesículas Extracelulares (ISEV) ha elaborado una guía con la información mínima que debe contener un estudio de EVs (MISEV). Esta guía incluye diversas cuestiones a tener en cuenta: desde cuáles son los factores preanalíticos del estudio, hasta las recomendaciones para llevar a cabo ensayos funcionales. Podéis encontrar el artículo en el siguiente enlace:
Fruto del trabajo conjunto de la ISEV y de la Sociedad Internacional para el Avance de la Citometría (ISAC), se ha publicado una guía más específica para los estudios que utilicen la Citometría de Flujo (CITF) como el método de elección para la caracterización fenotípica de EVs y su identificación y uso como biomarcadores:
Vamos a ver a continuación algunas cuestiones a tener en cuenta en un estudio de este tipo, incluidas en esta guía.
Fig. 2. Esquema de la información mínima que debe tener un estudio de EVs por citometría.
VARIABLES PREANALÍTICAS
Para trabajar con EVs va a ser necesario utilizar diversas técnicas que nos permitan confirmar que se ha realizado una correcta separación de las mismas a partir de una muestra biológica, así como identificar las moléculas contenidas en las mismas.
La elección de las técnicas más adecuadas para el enriquecimiento y caracterización de las EVs va a venir definida por el objetivo final de nuestra preparación, así como por la pregunta científica. Así, no es lo mismo querer determinar el cargo de las EVs (ya sea procedentes de un biofluido o de un medio condicionado), que identificar un determinado tipo de EV en un biofluido para usarlo como un biomarcador, ni tampoco caracterizar las EVs procedentes de un medio condicionado. Cada uno de los objetivos determinará tanto las variables preanalíticas a tener en cuenta como el método de enriquecimiento, purificación, caracterización y determinación del cargo.
Por ejemplo, en el caso de estudiar EVs procedentes de un medio condicionado, hay que tener en cuenta desde las placas o contenedores en que se cultivan dichas células, hasta la confluencia, tiempo de pase, procedimientos de aislamiento, nº de células muertas presentes en el cultivo, y algo muy importante: la composición del medio de cultivo y la utilización de sueroLIBREde EVs. Esto es así porque el suero fetal bovino es una fuente muy importante de EVs. Este tipo de suero se puede adquirir en una casa comercial específica, o podemos realizar un procedimiento para eliminar esas EVs mediante ultracentrifugación. En los siguientes enlaces se puede encontrar un protocolo de obtención de FBS libre de EVs, así como un ejemplo de protocolo de aislamiento por ultracentrifugación + gradiente:
En el caso de la obtención de EVs a partir de biofluidos, como por ejemplo de plasma, es muy importante estandarizar los métodos de obtención y transporte de la muestra, incluyendo el tipo de aguja utilizada para la extracción, el tipo de tubo usado para la recolección siendo el recomendado el de citrato sódico como anticoagulante, descartar los 2-3 primeros ml de sangre extraída, omitir muestras hemolizadas, y procesar las muestras lo antes posible, ya que los fluidos no procesados son inestables y pueden seguir produciéndose EVs por parte de las células presentes en la muestra. Además, hay que tener en cuenta que la utilización de métodos de ultracentrifugación de plasma sin otras técnicas adicionales (como la utilización de gradientes adicionales) puede co-aislar gotas lipídicas que contengan quilomicrones y LDL que puedan darnos datos erróneos en la cuantificación de EVs.
PREPARACIÓN DE LAS MUESTRAS
Una vez obtenidas muestras enriquecidas en EVs por cualquiera de los distintos métodos posibles (ultracentrifugación, filtración, cromatografía de exclusión molecular, etc), para caracterizarlas por CITF hay tres variables fundamentales:
Tinción de las muestras: puesto que las EVs son muy pequeñas, el nº de moléculas de anticuerpo que van a incorporar a su superficie va a ser muy pequeño con lo que vamos a necesitar anticuerpos y reactivos de calidad, con una conjugación de fluorocromo eficiente y de alto brillo.
Lavados: necesarios para eliminar el ruido del reactivo. Al aumentar el nº de lavados para eliminar el reactivo de tinción, pueden perderse EVs y alterar las características de las muestras en cuanto a composición de las EVs.
Diluciones: para una adecuada detección de las EVs por CITF va a ser necesario llegar a una concentración adecuada que nos permita detectar partículas individuales.
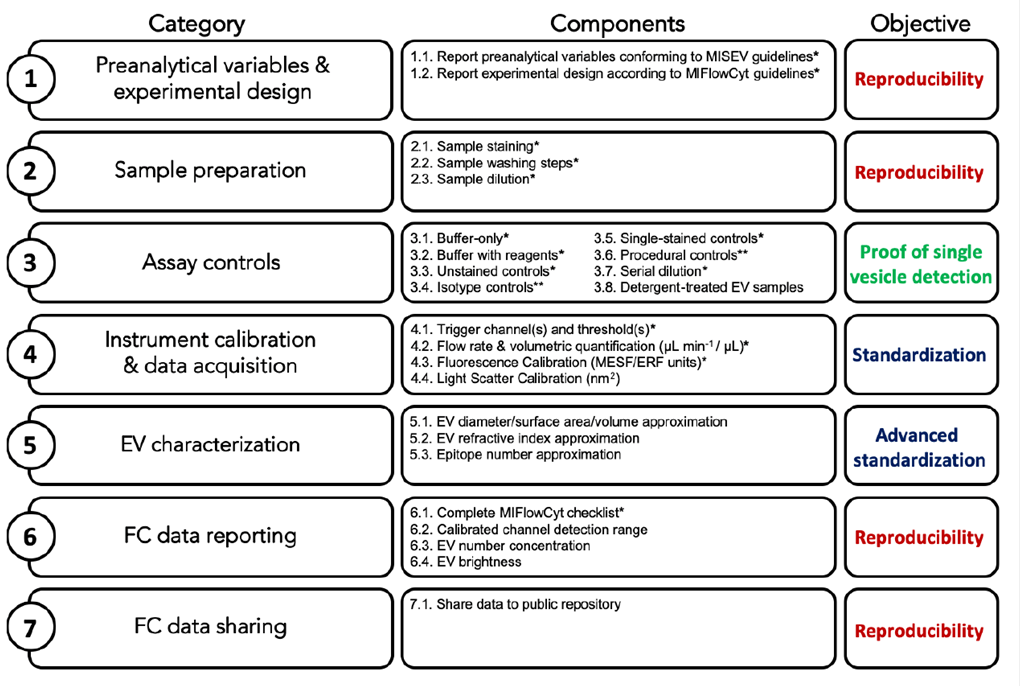
CONTROLES
Puesto que las EVs están en el límite de detección de los equipos, es muy importante establecer una serie de controles que nos permitan asegurarnos de que estamos identificando correctamente las EVs y de manera individual.
CONTROL SÓLO TAMPÓN: necesario para comprender cuáles son las señales de fondo producidas por el medio en el que se encuentran diluidas las EVsà adquisición del medio de dilución de las EVs
CONTROL TAMPÓN + REACTIVOS: Los reactivos no unidos pueden agregar y ser detectados como eventos positivos à adquirir muestra que contenga el medio de dilución de las EVs y los reactivos a la misma concentración utilizada para teñir las muestras. Deben analizarse con la misma dilución y los mismos settings experimentales que la muestra problema.
CONTROL SIN MARCAR: La finalidad es determinar si la muestra puede presentar autofluorescencia en los mismos canales en los que vamos a buscar las moléculas de interés à deben tener la misma concentración de EVs que la muestra teñida y ser analizadas con la misma dilución y los mismos settings experimentales que la muestra teñida.
CONTROLES DE ISOTIPO: La finalidad de estos controles es analizar la posible unión inespecífica de los anticuerpos utilizados en la muestraà muestras teñidas con anticuerpos del mismo isotipo y conjugados con los mismos fluorocromos que los anticuerpos con los que teñimos la muestra (a la misma concentración y con los mismos settings experimentales).
CONTROLES “SINGLE-STAINED”: Controles para identificar la contribución de cada fluorocromo a cada detector utilizado à muestras teñidas sólo con un anticuerpo, también usados como controles de compensación. En las mismas condiciones de la muestra teñida.
CONTROLES DE PROCEDIMIENTO: Algunos protocolos de tinción requieren procesamientos posteriores a la tinción à hay que realizar todos los controles que incluyan todas las variables del procedimiento.
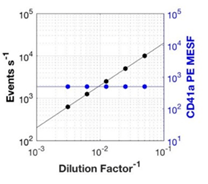
DILUCIONES SERIADAS: Estos controles de diferentes diluciones van a servir para asegurarse de que estamos analizando partículas únicas à Hay que hacer diluciones seriadas y representar el factor de dilución contra el total del nº de eventos adquiridos y la intensidad de fluorescencia y/o la dispersión de luz para demostrar que maximizamos la detección de EVs individuales. Si observamos que disminuye el nº de eventos con la dilución pero no encontramos disminución en la intensidad de fluorescencia, estamos detectando partículas individuales.
Fig.3. Representación del nº de eventos y de la intensidad media de fluorescencia frente al factor de dilución
CONTROL CON DETERGENTE: Como las EVs están rodeadas por una bicapa lipídica, el uso de detergentes puede ayudarnos a comprobar esta naturaleza y que la señal que observamos no se debe a agregados de anticuerpoà añadir detergente (ej. Tritón X-100, Tween-20) a las muestras teñidas y analizarlas como las muestras problema. Si desaparece el marcaje, nos dará la prueba de que la tinción que observamos en la muestra es realmente debido a las EVs.
CONCLUSIONES FINALES
Al estar en el límite de detección de los equipos existentes en el mercado, los estudios con EVs requieren de una serie de controles adicionales que no se requieren en estudios con células completas.
Las guías sobre cuál es la información mínima que se debe recoger en los estudios con EVs, nos aportan una ayuda inestimable para el diseño experimental y la recolección de resultados.
Cuando comenzamos a trabajar con roedores de laboratorio empezamos a aprender mucho sobre líneas de ratones que existen a disposición de los investigadores. Es un mundo lleno de posibilidades. Aprendemos que los ratones consanguíneos son una herramienta muy útil que nos permite disminuir el número de animales que utilizamos en nuestros experimentos debido a las características que presentan estas líneas:
Son isogénicos: Todos los individuos son genéticamente iguales.
Alto porcentaje de homocigosis (>90%).
Uniformidad fenotípica, ya que son genéticamente iguales.
Estabilidad genética a lo largo del tiempo.
Permite la comparación de resultados entre diferentes laboratorios a lo largo del tiempo.
¿PERO TODO ESTO ES REALMENTE ASÍ?
Miremos el ratón consanguíneo C57 BL6 tan utilizado como control en los estudios con ratones transgénicos que se mantiene en ese fondo de línea. El ratón C57BL6 de pelaje oscuro casi negro fue creado por Clarence Cook Little, fundador de The Jackson Laboratory.
C. C. Little se graduó en Harvard y comenzó su tesis en el laboratorio de Genética. Una de sus hipótesis de trabajo era que el cáncer tenía un fuerte componente hereditario y, para poder estudiarlo, necesitaba una cepa de ratones idénticos. Procreó entre sí los descendientes de una ratona que había en el laboratorio, hermana con hermano, durante veinte generaciones, y consiguió cepas en las que todos los individuos tenían la misma dotación genética y eran fértiles. Así, en 1927, obtiene la cepa C57BL/6.
Desde entonces se ha distribuido la cepa a cientos de Institutos y a miles de laboratorios en todo el mundo. Debido a la deriva genética, cada una de las subcepas está relacionada con la C57BL/6 pero contienen diferencias únicas, conocidas o desconocidas, en su secuencia genómica.
El patrimonio genético es un componente del diseño de los experimentos que puede afectar a su reproducibilidad y a la capacidad de hacer generalizaciones sobre los procesos biológicos.
La deriva genética es una realidad inevitable en las colonias de ratones, y puede tener un fuerte impacto sobre las conclusiones y la reproducibilidad de la investigación.
A la hora de trabajar con transgénicos hay que asegurarse que la nomenclatura es correcta para saber que subcepa de C57BL6 es la adecuada a utilizar como control. No todos los C57BL6 que distribuyen las casas comerciales son genéticamente iguales. Como hemos comentado antes, la elección de un control no adecuado puede llevar a conclusiones erróneas.
Por lo tanto, recomendamos usar los animales control con el mismo fondo genético en el que se mantiene el transgénico.
Algo en lo que insistimos en nuestros cursos de formación y que muchos alumnos nos preguntan es por la preparación de las muestras.
Los más veteranos ya sabréis la respuesta. No, no existe un protocolo perfecto o universal que funcione bien para todo tipo de muestras.
Las condiciones óptimas de preparación deben ser determinadas empíricamente para cada tipo de muestra. Dependiendo de la naturaleza de la muestra (tejido, fluido biológico, células), de la información que queramos obtener y de las técnicas que vayamos a utilizar para resolver nuestro problema biológico, adaptaremos unos métodos de separación, homogeneización, solubilización, limpieza, etc.
No he descubierto la pólvora ¿verdad?
Pues algo tan sencillo y que todos tenemos en nuestras rutinas, es el principal cuello de botella cuando se quiere realizar un análisis en proteómica. Elegir los métodos con los que obtengamos unos mejores rendimientos de extracción de proteínas con tampones compatibles a los protocolos de digestión y su posterior análisis por espectrometría de masas es fundamental para la generación de datos precisos e informativos.
Hoy en día, las tecnologías de última generación en proteómica permiten la identificación de miles de proteínas a partir de una pequeña cantidad de muestra, por ejemplo, clasificadas por FACS, microdiseccionadas con láser o células fijadas con formalina. Estos avances incluyen métodos mejorados de preparación de muestras, tecnologías de separación para llegar a una aproximación de proteómica unicelular basada en espectrometría de masas (MS). En la revisión de Lombard-Banek C. et al; 2020; podréis encontrar como los enfoques proteómicos se aplican en procesos de terapia celular.
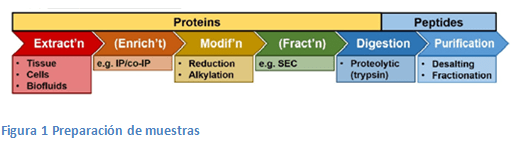
Recordemos como es el proceso de trabajo en proteómica
Primero, las muestras biológicas se preparan a través de un flujo de trabajo ascendente, en el que las proteínas se extraen de las células y se digieren enzimáticamente en péptidos. Los péptidos posteriormente se analizaran por LC-MS/MS.
Las aproximaciones que han surgido en los últimos años en el tratamiento de muestras tienden a que en un único vial se pueda realizar la gran mayoría de los pasos para aumentar el rendimiento del proceso, sobre todo cuando se parte de cantidades mínimas de muestra.
En la tabla se resume algunos de estos procedimientos y se comparan sus características.
FASP: Filter-aided simple preparation. La lisis de proteínas y la limpieza de péptidos se realizan en receptáculos separados en FASP, creando una fuente potencial de pérdidas de adsorción.
Pioneros en la mayoría de los avances en la preparación de muestras basadas en filtros generalmente usa filtros de corte de peso molecular (MWCO) que van desde 1 a 100 kDa de tamaño. La digestión enzimática se realiza en los propios filtros y los péptidos resultantes se liberan por centrifugación.
Ventajas: FASP mejoró las tasas de identificación de péptidos en comparación con el enfoque tradicional de digestión “in-solution”.
Inconveniente: FASP es un protocolo largo y requiere cantidades sustanciales de material.
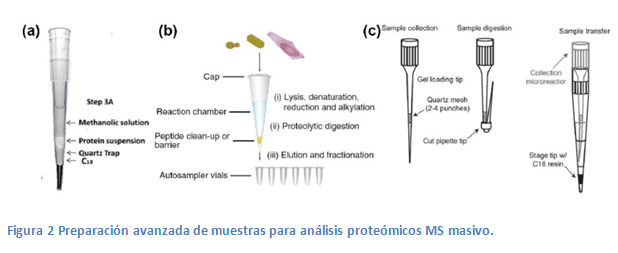
S-Trap: Suspensión trapping. Permite la limpieza, digestión y fraccionamiento de péptidos dentro de un solo dispositivo (Figura 2a). El dispositivo S-Trap está diseñado para un flujo de trabajo basado en la centrifugación y está compuesto por un filtro de cuarzo para atrapar proteínas intactas seguido de un tapón monolítico C18 (fase reversa) en el que se retienen los péptidos y se realiza la desalinización y / o fraccionamiento de la muestra.
Ventajas: El proceso en S-Trap se simplifica y es más rápido en comparación con el protocolo FASP debido a su diseño integrado. Se pueden preparar muestras con menos material proteico y el enfoque es compatible con una amplia gama de detergentes.
iST: In-Stage tip. Las células se pueden lisar directamente en el dispositivo (Figura 2b). El iST es simplemente una punta de pipeta con un inserto de disco C18 que sirve como barrera y dispositivo de limpieza de péptidos. Gracias al diseño de la punta, la preparación de múltiples muestras puede paralelizarse con el uso de pipetas multicanal o incluso automatizarse mediante dispositivos robóticos. De hecho, todo el procesamiento puede realizarse en un dispositivo de 96 pocillos, mejorando enormemente el rendimiento de la preparación de muestras.
Ventajas: En un estudio comparativo de iST con FASP, iST condujo a una mejor cobertura de proteínas de membrana y nucleares que FASP.
Inconveniente: iST no puede eliminar detergentes iónicos como SDS. Además, la muestra se calienta a alta temperatura antes de la digestión (> 60 ºC), por lo que debemos evitar agentes caotropicos como la urea.
Streamlined iST: Diseño combinado de S-Trap e iST. Para análisis proteómicos de células clasificadas por FACs (Myers, S.A. et al; 2019). Las células se recogen durante la clasificación directamente en la punta con el filtro de cuarzo quedando retenidas. La punta del filtro se dobla para evitar pérdidas durante la digestión y el pliegue se mantiene mediante un anillo cortado de una punta de pipeta. Las células se lisan y la proteína extraída se digiere (Figura 2c, en el centro). Luego se inserta la punta de preparación en una segunda punta que contiene un disco de C18 para la limpieza de péptidos (StageTip).
Ventajas: Reducción de la cantidad de partida de muestra, menos de 2 µg de contenido de proteína, lo que representa una disminución de ~ 5 a 100 en comparación con los enfoques tradicionales de EM (Tabla 1).
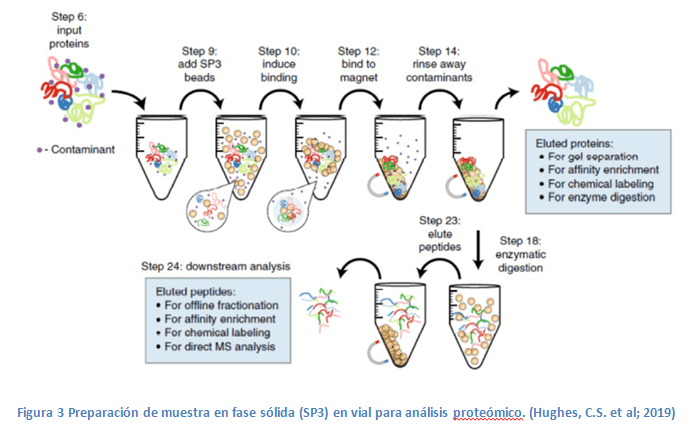
SP3: Single pot solid phase-enhanced simple preparation. Basado en el enfoque clásico de preparación de muestra en vial. Se utilizan bolitas mágneticas derivatizadas con grupos funcionales hidrofílicos (Figura 3). Este enfoque permite el enriquecimiento de proteínas, la eliminación de detergente y la digestión disolución al agregar las bolitas directamente en el lisado celular.
Ventajas: Todos los pasos del procesado de la muestra se realizan en un solo tubo y el costo del ensayo es menor que el de los enfoques descritos anteriormente. Este enfoque es compatible con una amplia gama de detergentes y agentes caotrópicos. Además, es compatible con un amplio intervalo de cantidad de partida de muestra, de muy baja a alta, al cambiar únicamente el tamaño del tubo de microcentrífuga utilizado y la cantidad de perlas magnéticas añadidas.
Bibliografía:
Lombard-Banek C, Schiel JE. Mass Spectrometry Advances and Perspectives for the Characterization of Emerging Adoptive Cell Therapies. Molecules. 2020; 25(6):1396. Published 2020 Mar 19. doi:10.3390/molecules25061396 https://pubmed.ncbi.nlm.nih.gov/32204371/
Sielaff M, Kuharev J, Bohn T, et al. Evaluation of FASP, SP3, and iST Protocols for Proteomic Sample Preparation in the Low Microgram Range. J Proteome Res. 2017; 16(11):4060‐4072. doi:10.1021/acs.jproteome.7b00433
Pieragostino D, Lanuti P, Cicalini I, et al. Proteomics characterization of extracellular vesicles sorted by flow cytometry reveals a disease-specific molecular cross-talk from cerebrospinal fluid and tears in multiple sclerosis. J Proteomics. 2019;204:103403. doi:10.1016/j.jprot.2019.103403
M. Rezaa Mohammadi, Milad Riazifar, Egest J. Pone, Ashish Yeri, Kendall Van Keuren-Jensen, Cecilia Lässer, Jan Lotvall, Weian Zhao, Isolation and characterization of microvesicles from mesenchymal stem cells, Methods, Volume 177, 2020, Pages 50-57, ISSN 1046-2023, https://doi.org/10.1016/j.ymeth.2019.10.010
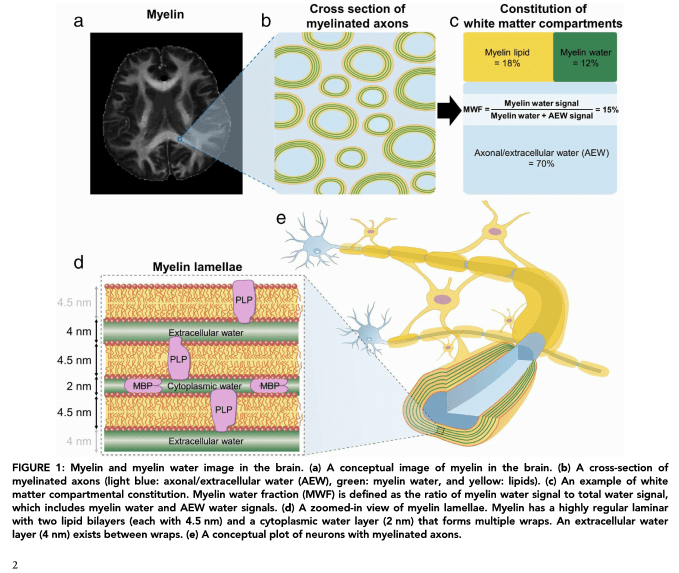
Empezamos la sección de la Newsletter del SAI RM tratando un tema de especial interés en vuestras líneas de investigación: ¿sería posible detectar la Mielina por Imagen de Resonancia Magnética? La respuesta es SI pero no con los técnicas de imagen convencionales, requiere de secuencias especiales y un análisis de imagen posterior.
En este primer artículo, me gustaría comentaros esta modalidad como una breve introducción, teniendo en cuenta que no todos vosotros estáis familiarizados con la técnica de RM. Trataré en los siguientes capítulos de ir explicando distintas aplicaciones e introduciendo en cada uno de ellas los parámetros más importantes. He tratado de dar unas breves notas, pues veréis, que hay una gran variedad tanto en adquisición como en el procesamiento de la imagen. Al contrario de lo que ocurre en clínica, se pueden modificar muchos parámetros, pero al igual que esto trae sus ventajas también trae sus desventajas. Se abre un mundo de posibilidades en los que simplemente la modificación de un solo parámetro te hará variar tanto la imagen (contraste, resolución, etc… ) como el tratamiento de la imagen con el postprocesado. Las figuras y tablas que muestro están extraídas de diferentes artículos como muestra y ejemplo de datos que podríamos obtener. Lo ilustro a modo de ejemplo, y como base para abrir ciertas inquietudes…. Empecemos:
La imagen de Resonancia Magnética está basada principalmente en la señal que producen los protones del agua y de distintos componentes que forman las proteínas y los lípidos. Si bien, es posible detectar otros núcleos empleando antenas de radiofrecuencia específicas, prácticamente la mayoría del diagnóstico clínico e imagen preclínica está basada en la imagen de los protones del agua.
En el sistema nervioso (así como en otros tejidos) los protones se encuentran formando parte de diversas estructuras que son detectables y caracterizables por IRM teniendo en cuenta los parámetros de relajación que varían en función del campo magnético.
Concretamente, el tejido nervioso mielinizado está compuesto por varios componentes que tienen distintas propiedades magnéticas (distintos tiempos de relajación). La señal que producen los protones en las diferentes estructuras mielinizadas es apenas detectable porque tienen valores de T2 muy cortos, (rango de microsegundos) empleando secuencias de imagen de RM convencionales. Es más, estos componentes se suelen omitir en los modelos teóricos a partir de los datos de la imagen.
Debido a estos tiempos de relajación tan cortos, se han ido desarrollando secuencias que detecten la mielina indirectamente, estudiando las propiedades del agua asociada a las vainas de mielina y sus interacciones con los protones asociados con proteínas y lípidos que forman éstas.
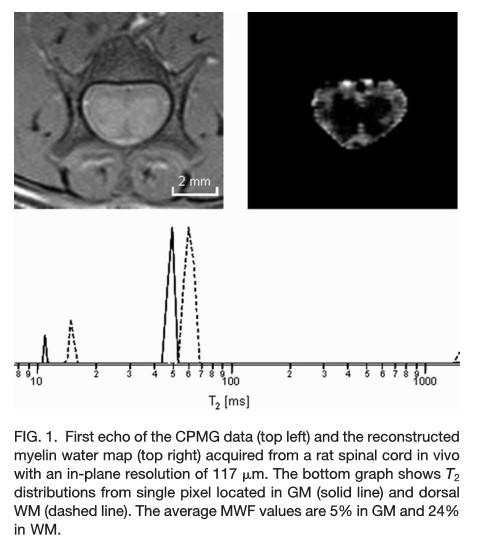
Una de las técnicas llamada MWI (Myelin Water Imaging) va a estudiar las diferencias en los tiempos de relajación T2 de los protones del agua cuando ésta se encuentra formando parte de distintas estructuras.
Se pueden encontrar 3 componentes que contribuyen a esta señal:
En el rango de milisegundos agua unida a mielina: representa la MWF (Myelin Water Fraction)
Un segundo componente en el rango de 50-100 ms que corresponde al agua que se encuentra intra-extracelular (I/E) denominada “other water fraction OWF”
Y un tercer componente de varios cientos de milisegundos que representa el líquido cerebroespinal (agua libre)
La siguiente figura representa el rango de T2 de los dos primeros componentes: teniendo en cuenta estas dos señales, se puede calcular la fracción de Mielina-agua
De esta manera se podrá calcular la fracción Mielina-agua (MWF) que representará la cantidad de agua atrapada entre la bicapa de Mielinia como la representación de las curvas de decaimiento multiexponencial de los componentes de T2 cortos obtenidas de las imágenes multieco.
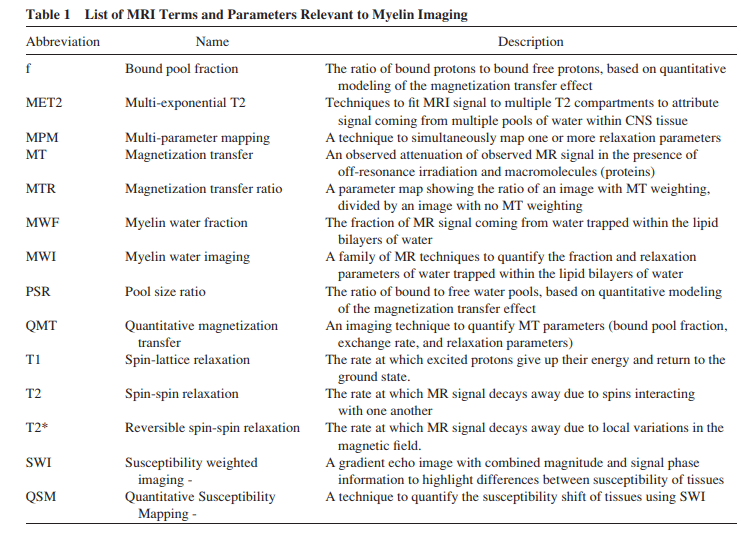
Para que podáis estar más familiarizados con la terminología empleada, la siguiente tabla describe estos parámetros y sus principales características:
En la literatura podemos encontrar varias metodologías que estudian un intercambio de protones de agua libre con la ligada a macromoléculas. Es el caso de la técnica de Transferencia de Magnetización. Esta técnica que aplica un pulso de saturación a una frecuencia de resonancia del espectro determinada, no es específica de los protones unidos a mielina. Es decir, estaríamos produciendo un cambio en el contraste, un cambio “cuantitativo” (en los mapas procesados) pero no podríamos asegurar que este cambio estuviese producido solamente por la Mielina.
En las siguientes gráficas se muestra el distinto comportamiento de la señal del agua unida a Mielina y el agua extracelular/axonal con respecto a los valores de T2, T1 y Transferencia de Magnetización.
Si bien, “parece” relativamente fácil implementar la secuencia, reproduciendo los parámetros que se detallan en los artículos (teniendo en cuenta: el campo magnético, las antenas de radiofrecuencia y gradientes del equipo empleado, etc.) es en el post-proceso de las imágenes donde se requiere más conocimiento y dedicación.
Y ya, para terminar, una mención a la imagen de la Mielina en médula. A parte de los estudios de difusión que aportan muchísima información sobre cómo están orientadas las fibras. Podríamos trabajar con la MWI como secuencia complementaria, teniendo en cuenta una serie de inconvenientes:
Problemas de susceptibilidad magnética, nos encontramos en un medio no homogéneo, mucha interfase tejido medular-tejido óseo
Inhomogeneidad del campo, es muy difícil obtener un shimming correcto con tanta interfase y artefacto de respiración y flujo
Presencia de líquido cerebroespinal cuyos valores de T2 podrían enmascarar los valores de T2 buscados
Programar las secuencias disponibles en el equipo y ajustar los valores de adquisición requeridos: secuencias multieco y secuencias UTE (ultrashot time echo); donde a veces el ajuste de estos parámetros se ven comprometidos por la configuración del equipo.
La manera de obtener imágenes tridimensionales de la médula a buena resolución y en poco tiempo.
En la newsletter de hoy el SMAI quiere presentaros la microscopía de fluorescencia de lámina de luz o LSFM acrónimo de la técnica en inglés (Light sheet fluorescent microscopy). Esta es una técnica de microscopía de fluorescencia con una buena resolución óptica en XY y con muy buena resolución en Z además de ser de alta velocidad, lo que permite ser la mejor técnica para seccionamiento óptico (reconstrucciones 3D).
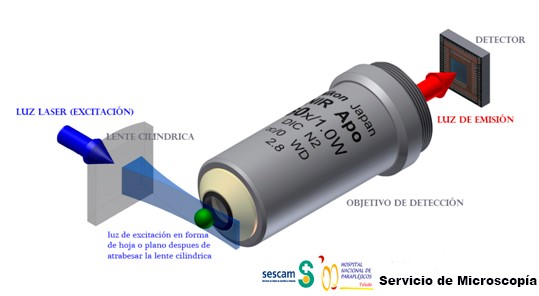
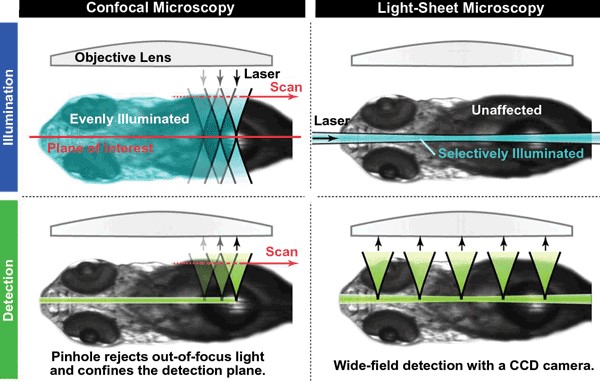
LSFM es un microscopio de fluorescenca, pero con una diferencia muy importante con la microscopía de fluorescencia convencional y la confocal, esta diferencia, es la forma en la que se ilumina la muestra. Mientras que en los microscopios de fluorescencia convencional (epifluorescencia) la iluminación llega atreves del objetivo para excita la muestra y la luz emitida por dicha muestra es recogida de nuevo por el objetivo, en la LSFM la muestra se ilumina perpendicularmente a la observación de la muestra. Esta iluminación perpendicular a la observación permite iluminar solo una porción muy delgada (generalmente de unos cientos de nanómetros a unos pocos micrómetros) de la muestra (ver figura 1).
Para la iluminación, se utiliza una lámina de luz láser, es decir, un rayo láser que se enfoca solo en una dirección creando un plano de luz (por ejemplo, usando una lente cilíndrica). Como solo se ilumina la sección realmente observada, este método reduce el fotodaño y el estrés producido a muestras vivas. Además, la buena capacidad de seccionamiento óptico reduce la señal de fondo y, por lo tanto, crea imágenes con mayor contraste, comparable a la microscopía confocal. Debido a que LSFM escanea muestras usando un plano de luz en lugar de un punto, puede adquirir imágenes a velocidades de 100 a 1000 veces más rápidas que las que ofrecen los métodos de microscopía confocal que escanean punto a punto toda la muestra.
En la actualidad varias casas comerciales venden microscopios para hacer LSFM y para ello crean unas cámaras donde colocar la muestra a observar. Esas cámaras están compuestas por juegos de dobles objetivos, uno para el sistema de iluminación y otro objetivo para la observación o detección. El objetivo para la observación es un objetivo convencional con sus lentes correspondientes y el objetivo de iluminación es perpendicular al anterior y contiene una lente cilíndrica que crea el plano u hoja de luz que ilumina la muestra (ver figura 2 donde en el primer cuadrante “A” se muestra un sistema normal de epifluorescencia y en el resto de cuadrantes se muestran diferentes configuraciones de la cámara de observación e iluminación para LSFM).
Para entender bien esta técnica debemos compararla con Microscopia confocal y con microscopia convencional de WF (campo amplio, microscopio de epifluorescencia o los VTL que tenemos en el servicio). La microcroscopia de WF es mucho más rápida que la microscopía confocal, puesto que se recoge todo el campo observado con un detector (CCD) y el tiempo de captura solo depende del tiempo de exposición. La microscopía confocal es mucho más lenta puesto que la muestra es escaneada punto a punto por la luz laser y la imagen ha de formarse después del escaneo. Sin embargo la resolución en Z es mucho mejor en la microscopía confoca, permitiéndonos hacer reconstrucciones 3D, gracias al pinhole de la microscopía confocal solo detectamos la emisión de la muestra de un plano focal muy delgado, quitando de la imagen la zona desenfocada por encima y por debajo del plano focal. Por tanto, mientras que con microscopia de campo amplio (WF) es más rápida, la imagen no nos permite hacer reconstrucciones 3D ni ver en detalle los planos en Z, puesto que nos muestra todo lo excitado en profundidad, incluso aquello que está desenfocado. (ver figura 3, comparación microscopia WF y microscopía confocal).
Pues bien, la técnica de LSFM combina la velocidad de WF con la resolución en Z de la microscopia confocal, pero además a la hora de excitar la muestra y gracias a la iluminación en forma de plano u hoja de luz muy delgada, solo es excitado el Z capturado, ahorrándonos el floto daño que se produce tanto en la microscopia WF como en la confocal.
En la siguiente imagen comparamos tanto la iluminación como la detección en la microscopía confocal con la LSFM.
En la parte izquierda vemos como funciona la iluminación (arriba) y la detección (abajo) del microscopio confocal. Vemos que se ilumina gran parte de la muestra produciéndose un escaneo punto a punto y se detecta un plano de interés muy delgado pero punto a punto. En la parte derecha (LSFM) vemos que la muestra solo es iluminada en un plano delgado y se detecta a la vez todo el plano iluminado. Moviendo esa iluminación de abajo arriba podemos hacer una reconstrucción 3D de toda la muestra en un tiempo relativamente rápido y sin producir un daño importante por la luz.
Con todo esto, esta técnica es muy útil para trabajar en biología celular e histológica. Principalmente se ha utilizado para el estudio de desarrollo embrionario en las primeras fases, haciendo videos de reconstrucciones 3D de los procesos del desarrollo con enorme resolución. Aquí tenéis un video muy corto de cómo quedaría en las primeras fases de desarrollo embrionario.
Pero, ¿cómo puede ser útil en las investigaciones que se hacen en el HNP?, Pues bien, esta técnica también se ha utilizado para el estudio de tejidos y órganos e incluso pequeños animales, pero para ello es necesario aclarar el tejido. En Neurociencia se ha utilizado en medula espinal sanas y lesionadas, pero siempre es necesario un proceso de aclarado con Tetrafurano (THF) del tejido. A continuación tenéis el enlace a dos artículos de los mejores que he visto sobre el aclarado de medula espinal y posterior utilización de LSFM.
Creemos que esta técnica puede ser muy útil para algunos de los grupos de la Udi del HNP y hemos investigado si sería posible acoplar este sistema de iluminación a alguno de los microscopios que tenemos, en principio parece que no es posible, pero en Ciudad Real compraron un equipo de LSFM el año pasado y podrían darnos servicio si fueses necesario, y quién sabe si en un futuro puede ser nuestra nueva adquisición.
The COVID-19 pandemic is an unprecedented global challenge. Highly variable in its presentation, spread and clinical outcome, novel point-of-care diagnostic classifiers are urgently required. Here, we describe a set of COVID-19 clinical classifiers discovered using a newly designed low-cost high throughput mass spectrometry-based platform. Introducing a new sample preparation pipeline coupled with short-gradient high-flow liquid chromatography and mass spectrometry, our methodology facilitates clinical implementation and increases sample throughput and quantification precision. Providing a rapid assessment of serum or plasma samples at scale, we report 27 biomarkers that distinguish mild and severe forms of COVID-19, of which some may have potential as therapeutic targets. These proteins highlight the role of complement factors, the coagulation system, inflammation modulators as well as pro-inflammatory signalling upstream and downstream of Interleukin 6. Application of novel methodologies hence transforms proteomics from a research tool into a rapid-response, clinically actionable technology adaptable to infectious outbreaks.
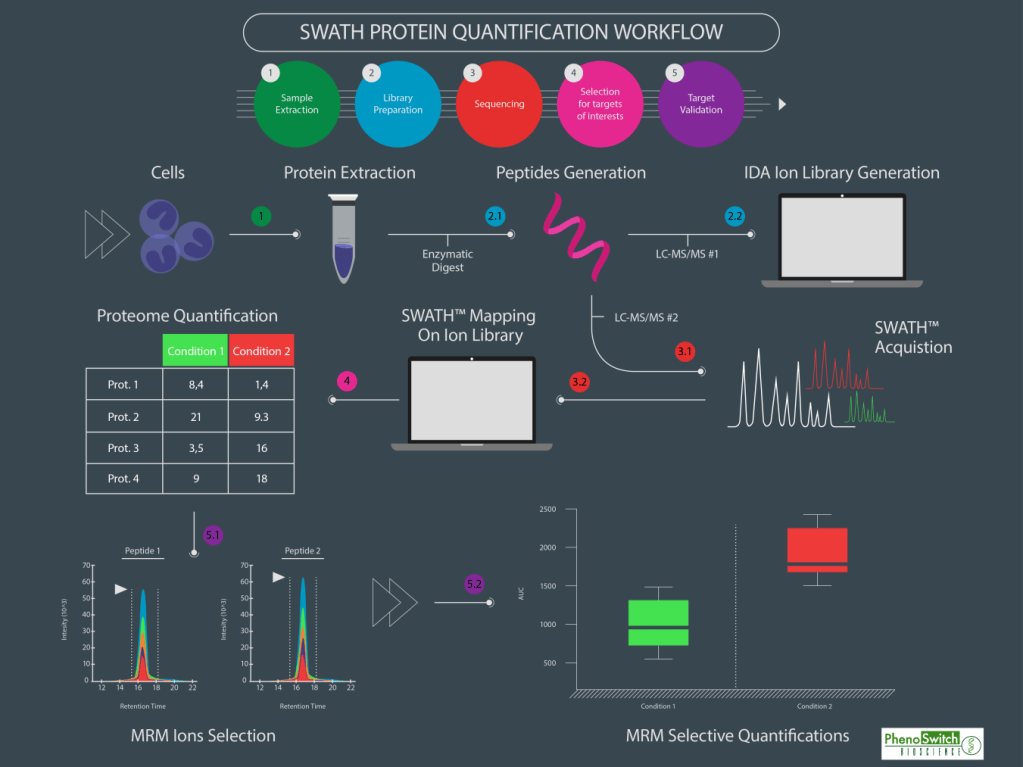
SWATH-MS® (Sequential Window Acquisition of All Theoretical Fragment Ion Mass Spectra)
Es un método de adquisición que genera mapas de proteínas globales y cuantitativas utilizando la combinación de la adquisición de datos independientes (DIA) y el análisis de datos específicos, de esta forma, aumenta enormemente el rendimiento de identificación/cuantificación de péptidos en comparación con el ya conocido SRM.
Antes de nada, un pequeño repaso de algunos términos:
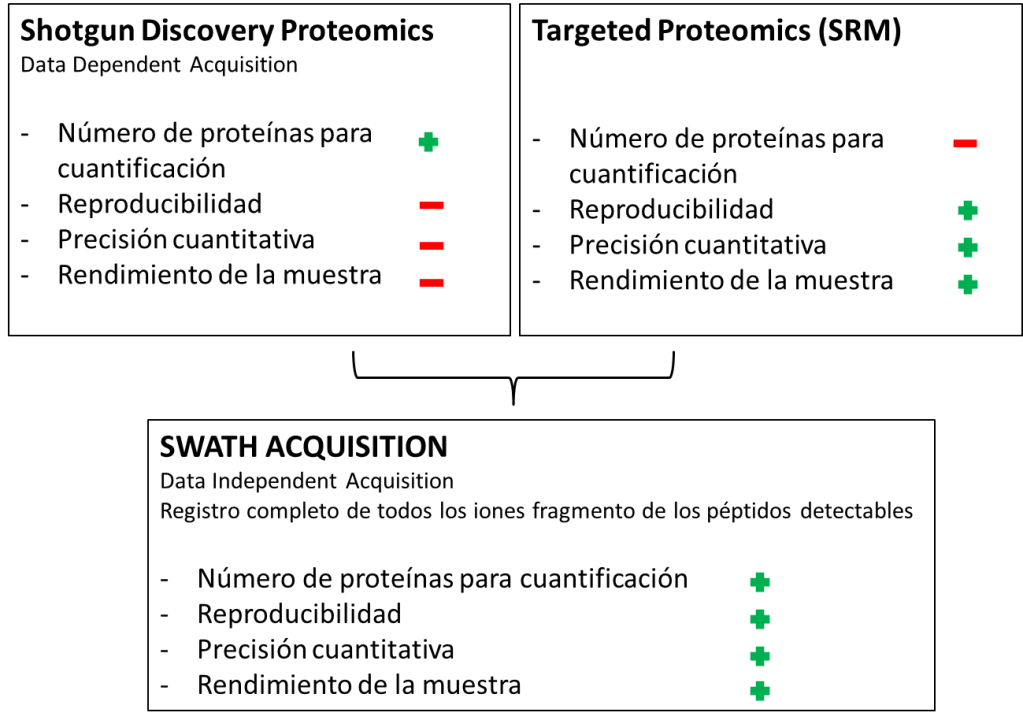
DDA (Data-Dependent Adquisition): También conocido como “shotgun proteomics”. Técnica llevada a cabo en espectrómetros tipo triple cuadrupolo (QqQ), de baja resolución; o en espectrómetros de alta resolución tipo Orbitrap, TripleTOF, QqTOF o MALDI-TOF/TOF. Todos los péptidos dentro de un cierto intervalo de masas son fragmentados.
En esta técnica se necesita de información previa para que el espectrómetro de masas seleccione los iones precursores más intensos y los fragmente.
Técnica con un número de iones precursor a fragmentar limitado dependiente de cada equipo.
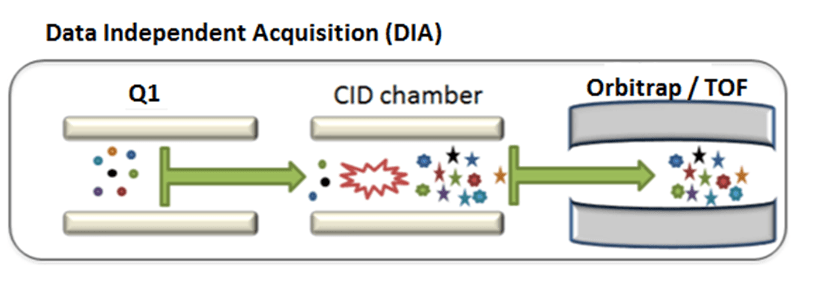
DIA (Data-Independent Adquisition): Todos los iones dentro de un intervalo de masa (m/z) son fragmentados y analizados. Técnica llevada a cabo en espectrómetros de masas de alta resolución con analizadores tipo Orbitrap, TripleTOF o QqTOF.
Técnica donde no es necesario conocer de antemano ninguna información.
SRM/MRM (Selected Reaction Monitoring/Multiple Reaction Monitoring): También conocido como “targeted proteomics”. Técnica llevada a cabo en espectrómetros de masas con analizadores de tipo triple cuadrupolo (QqQ), donde un ión de una masa particular es seleccionado en el primer cuadrupolo, fragmentado en el segundo cuadrupolo y uno de los productos de fragmentación es filtrado en el tercer cuadrupolo.
Técnica donde es necesario conocer la información del ion precursor y el ion fragmento a seleccionar de antemano.
Cada modo de adquisición tiene sus ventajas y desventajas. Dependiendo del tipo de muestra o tipo de experimento a llevar a cabo convendrá uno u otro.
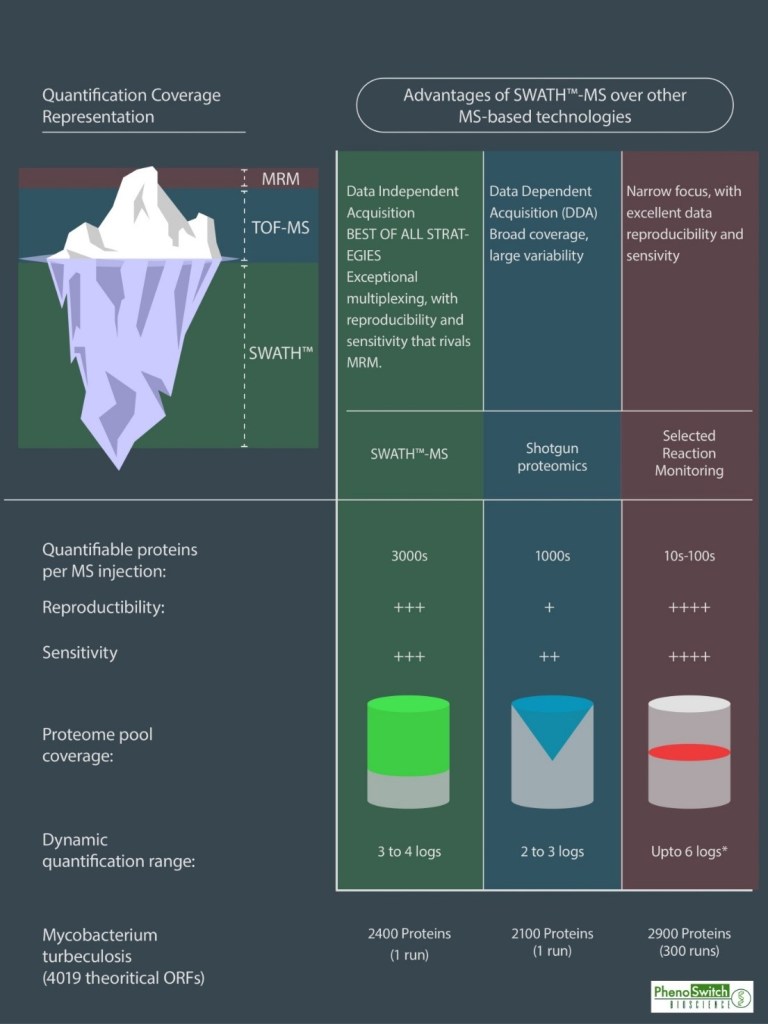
Pero la tendencia en los últimos años en proteómica es ir hacia análisis tipo DIA o SWATH donde la cobertura en adquisición y precisión es inmensamente favorable.
En este esquema se muestra gráficamente la cobertura de cada metodología, siendo el análisis SWATH-MS el más amplio con diferencia.
Previo al análisis SWATH es necesaria la creación de una librería de datos (cuanto más completa mejor) con la que se comparan los datos adquiridos en modo SWATH. Esta adquisición tiene la gran ventaja de que podrá ser utilizada para realizar una cuantificación además de poder ser revisada para distintas proteínas en un futuro sin la necesidad de re-analizar la muestra.
Aquí os dejamos un vídeo de SCIEX, creador de esta nueva tecnología: