El high content analysis (HCA) es una técnica instrumental que emplea microscopía automatizada en combinación con análisis de imagen para obtener un gran número de datos del estudio realizado. En nuestro centro, esta técnica se emplea principalmente para el estudio cuantitativo de eventos celulares complejos. La última newsletter de nuestro servicio (3 Mayo 2021, Javier Mazario) explica como identificar el número de células mediante la segmentación de sus núcleos para realizar HCA. En la newsletter de hoy explicaremos una variante de ese proceso diseñada por el SMAI del HNP para contar células con núcleos complejos (células con núcleos raros).
Mediante HCA podemos capturar de forma automática gran cantidad de fotos de un mismo tratamiento (pocillo de placa en cultivo) y procesarlas para contar los núcleos y por tanto las células, para posteriormente analizar los diferentes eventos contados y obtener gran cantidad de datos de esos análisis. Mediante diferentes pasos de pre-procesado, segmentación y post-procesado podemos obtener el número total de núcleos presentes en las fotos capturadas automáticamente. Debido a la variabilidad de las muestras y a las diferentes intensidades de marcaje de los núcleos puede que no identifiquemos todos o que se solapen varios si están muy próximos. Este problema lo solucionamos gracias al módulo de análisis de imagen por inteligencia artificial con el que cuenta el programa informático ScanR de nuestro nuevo equipo Olympus para HCA. Con él, podemos entrenar una red neuronal convolucional que aprenda a identificar los núcleos tal y como queda explicado en la newsletter anterior (3 Mayo 2021, Javier Mazario).
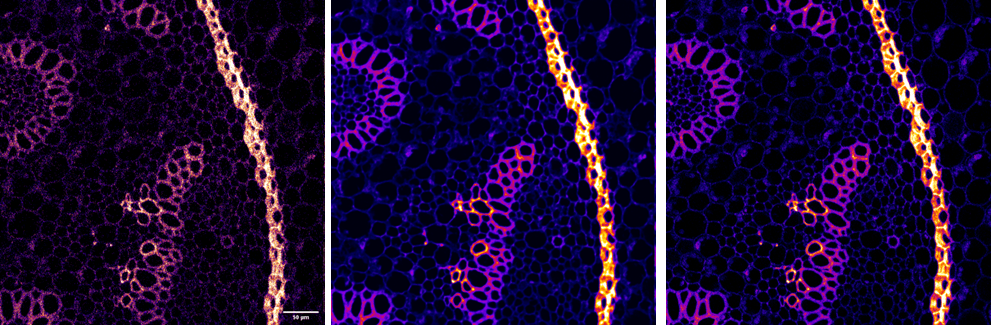
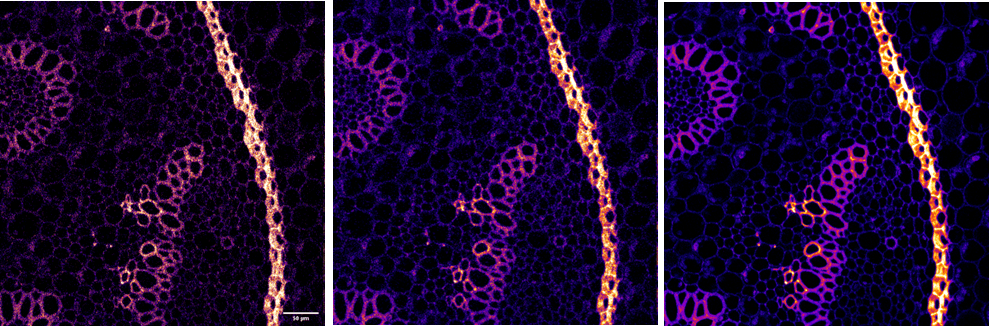
En la Unidad de Investigación del HNP, algunos grupos estudian diferentes fármacos que pueden afectar a la proliferación, diferenciación o migración de las células. El caso que nos ocupa hoy, es el de un fármaco antimitótico, que además de reducir la división celular, provoca que un alto porcentaje de las células que crecen en cultivo con ese fármaco, desarrollen núcleos raros. Aparecen células plurinucleares o células con núcleos cuyas formas son heterogéneas y nada convencionales (Figura1B). ¿Qué ocurre cuando tenemos gran cantidad de núcleos raros?, ¿cómo son identificados estos núcleos que se salen de lo habitual? Normalmente, este tipo de núcleos es poco habitual, pero debido a tratamientos antimitóticos específicos no es tan raro, más bien es la norma en los pocillos con el tratamiento.
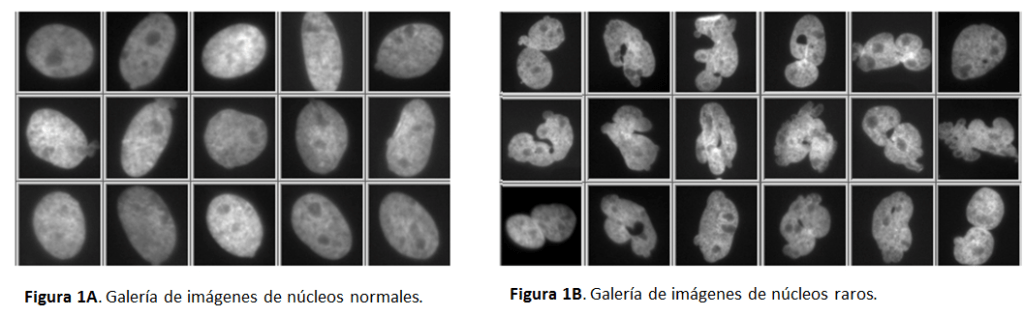
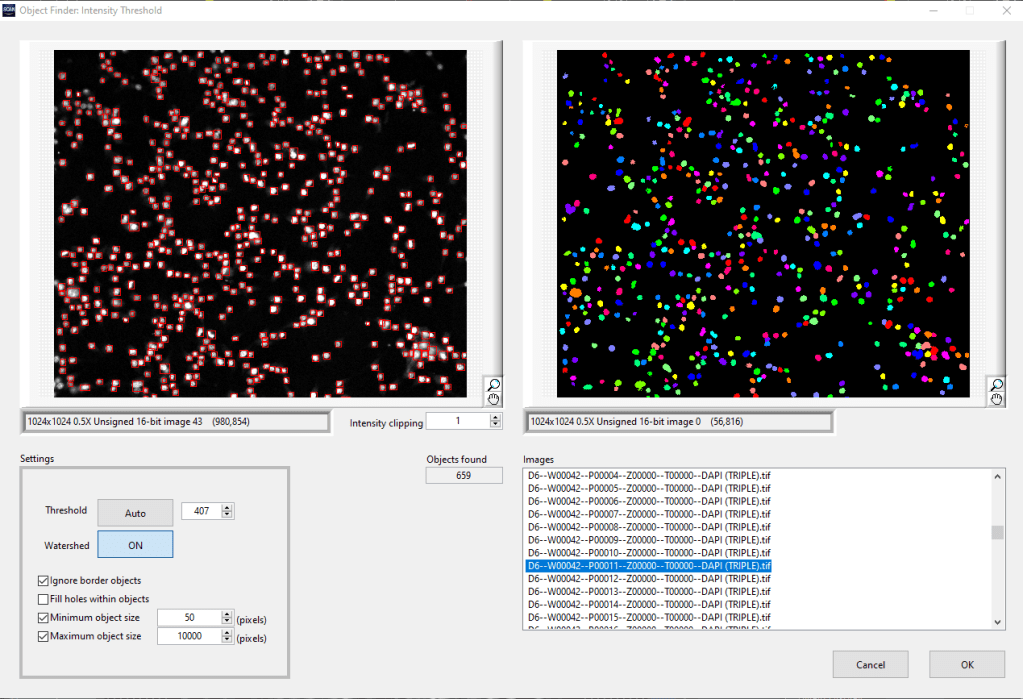
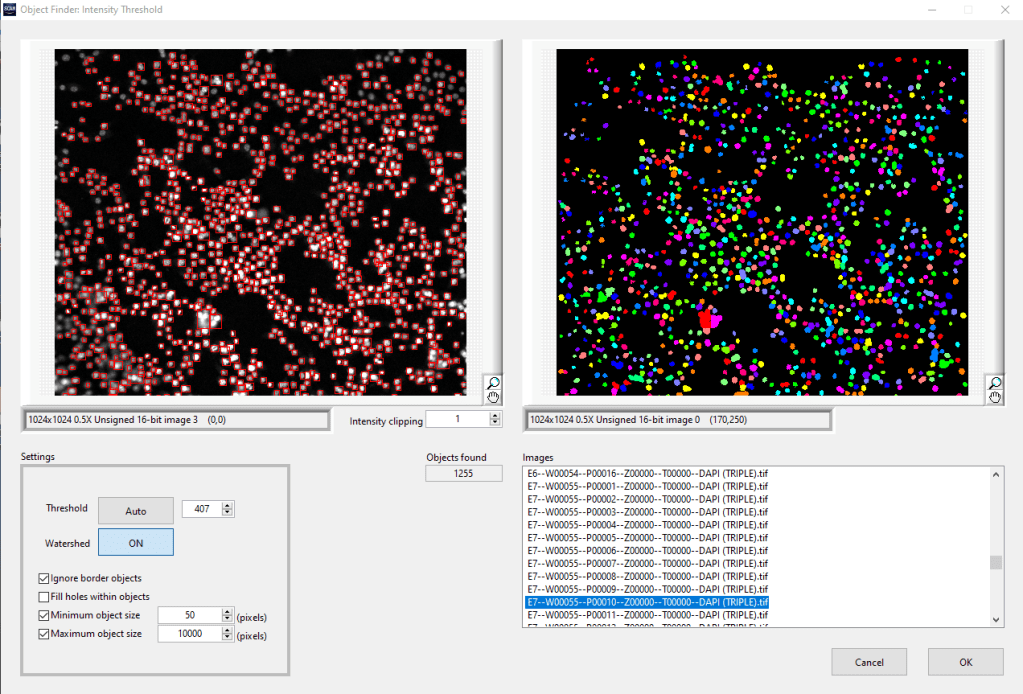
Figura 1. El software con el que hacemos HCA (SCANR de Olympus), nos permite hacer galerías de imágenes de los eventos seleccionados dentro de una región previamente definida con unas características concretas. En este caso los eventos son los núcleos de las diferentes fotografías de los pocillos de la placa de cultivo y las regiones están definidas por parámetros de tamaño y circularidad de los núcleos, identificando como núcleos raros aquellos de mayor tamaño y valores de circularidad alejados de la media.
Los núcleos raros presentan un problema a la hora de ser identificados con una red neuronal entrenada como se explicó en la Newsletter anterior (3 Mayo 2021, Javier Mazario). El problema está después de la segmentación, en el post-procesado de tipo watershed (Ver nota watershed a continucion).Este post-procesado separa núcleos adyacentes incorrectamente identificados como uno solo (Figura 2.A). Pero este tipo de post-procesado comete muchos errores cuando se trata de identificar núcleos raros ya que los trata como núcleos adyacentes y no como núcleos con formas diferentes a lo habitual (Figura 2.B).
NOTA (post-procesado de tipo watershed); La transformada Watershed es una técnica morfológica de segmentación de imágenes de niveles de grises. El concepto de Watershed procede del campo de la topografía. La imagen es interpretada como un relieve topográfico, donde se interpretan los diferentes niveles de grises como alturas. Las líneas Watershed son las fronteras de separación entre las cuencas de deyección de ríos y lagos. Además, cada cuenca está asociada a un mínimo local del relieve. De manera que utiliza esta técnica para identificar la frontera entre eventos (en nuestro caso núcleos).

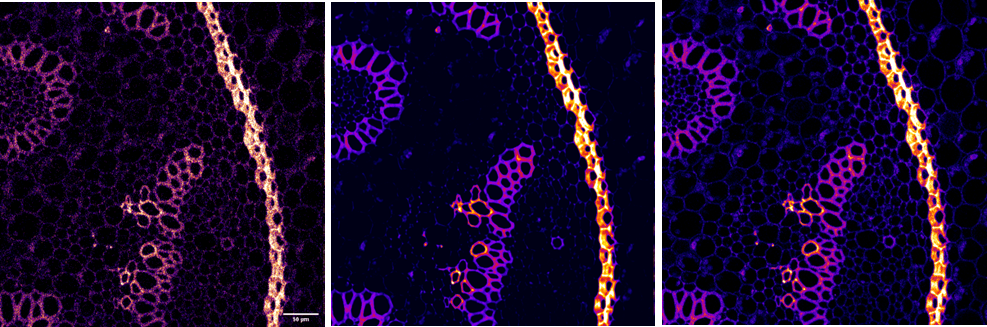
Figura 2A. Después de la segmentación sin post-procesado tipo watershed se identifican 4 núcleos, ya que no distingue dos de ellos que están muy próximos (arriba). Pero si se utiliza post-procesamos con watershed distingue los 5 núcleos reales (abajo).
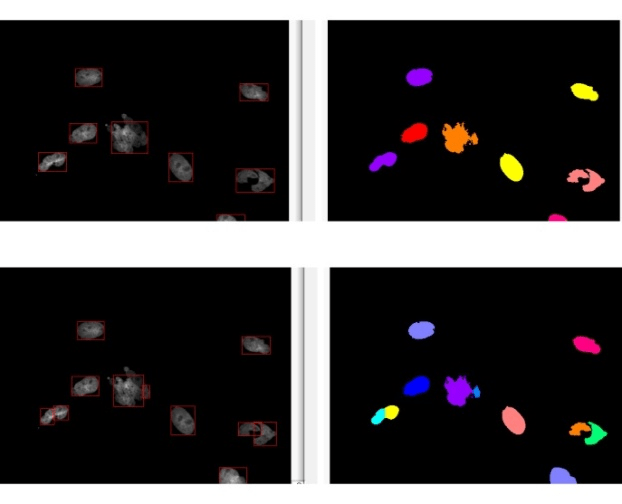
Figura 2B. Pero lo que es correcto en las fotos de los pocillos sin tratamiento (núcleos normales, figura 2A), resulta un problema en las fotos de los pocillos con tratamiento antimitótico (núcleos raros). En este caso el post-procesado tipo watershed separa como núcleos diferentes partes de un mismo núcleo (abajo) contando 11 células. En este caso lo útil seria no hacer el post-procesado, de manera que se identifiquen 8 células correctamente y no 11 (arriba). Pero al no hacer el post-procesado tipo watershed asumimos el error de contar como un mismo núcleo aquéllos núcleos adyacentes o muy próximos entre sí.
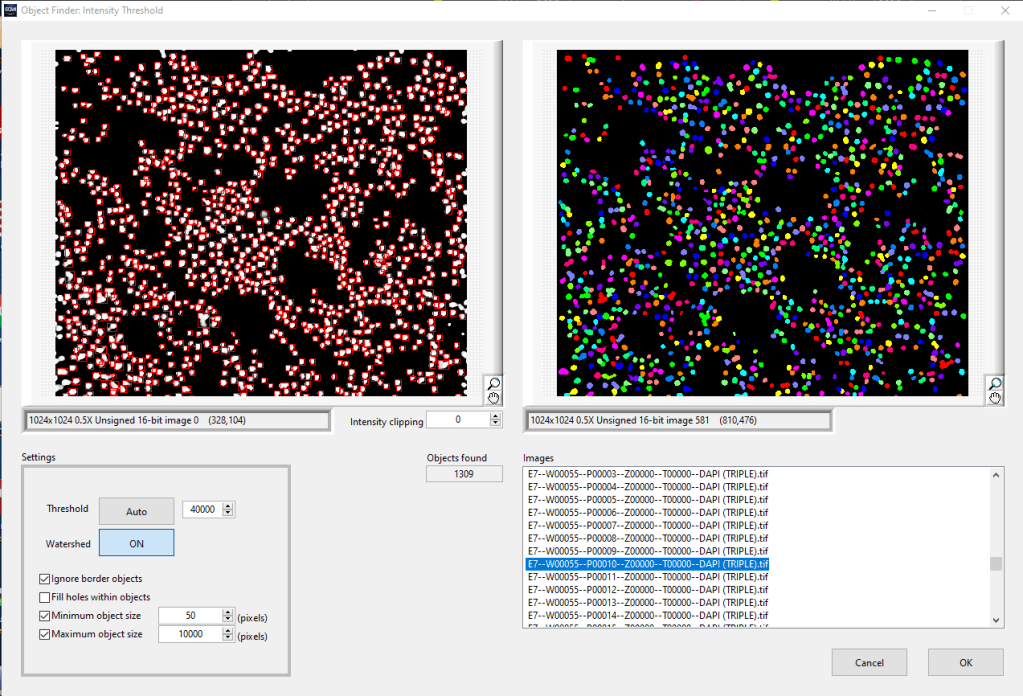
Para poder distinguir núcleos raros y además, separar los núcleos adyacentes de células diferentes, debemos enseñar a nuestro software de una manera más fina a identificar este tipo de núcleos. Para eso contamos con otra herramienta, el Software Cellsens de Olympus en el que nos permite hacer deep learning (aprendizaje profundo de la red neuronal), a partir de fotografías que le mostramos donde previamente hemos identificado (dibujado) lo que nos interesa contar.
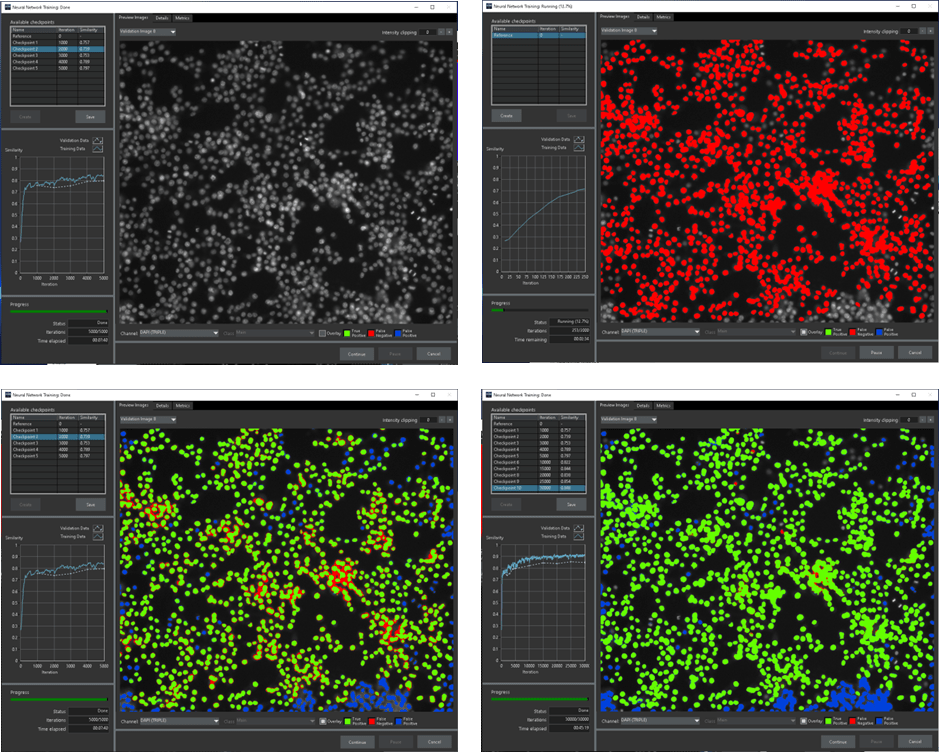
Este sería el mismo proceso que seguimos para procesar las imágenes con inteligencia artificial (AI) donde las imágenes que utilizaremos para enseñar al software tienen eventos difíciles de segmentar automáticamente (ver figura 3).
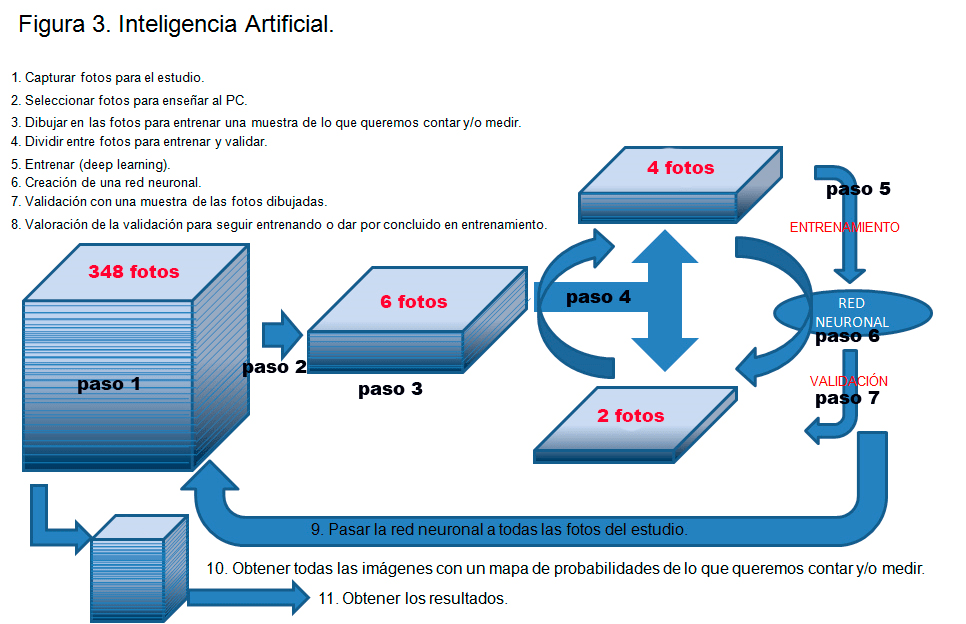
En la figura 3 mostramos un esquema de los pasos a seguir para entrenar, crear una red neuronal, validar dicha red y procesar las fotos con la red neuronal creada para obtener los resultados. En futuras Newsletter nos centraremos en los diferentes pasos a seguir para realizar este proceso. En esta Newsletter introducimos este esquema explicando brevemente algunos pasos que se realizaron para la identificación de los núcleos raros.
En el paso 2 se seleccionaron 4 fotos de los tratamientos antimitóticos donde el porcentaje de núcleos raros es alto y 2 fotos de los pocillos sin tratamiento donde todos los núcleos son normales. De las 6 fotos seleccionadas (figura3, paso 2) se hizo una segmentación automática y se post-procesaron con watershed. Pero tras el post-procesado se revisaron una a una las 6 fotos, borrando la máscara de segmentación donde se producían los errores visto en la figura 2B (abajo). Es decir, se borraban las máscaras de segmentación donde un núcleo raro aparecía como varios núcleos. Posteriormente se dibujó a mano los perfiles de núcleos raros donde se habían cometido los errores (figura 3, paso 3). Con esas fotos donde todos y cada uno de los perfiles nucleares estaban bien dibujados se entrenó una red neuronal mediante deep learning. Una vez validada esa red neuronal con altos valores de coeficiente de validación se dio por concluido el entrenamiento. Con la red neuronal obtenida se procesaron las 348 fotos capturadas automáticamente con el equipo de HCA (16 fotos por pocillo, 9 pocillos con tratamiento y 9 pocillos control, ver figura 4). Así obtenemos el número real de núcleos y por tanto de células en cada foto. El sumatorio de los núcleos de las 16 fotos de cada pocillo nos permite tener una idea del número de células totales por pocillo y por tanto como afecta el tratamiento al número total de células.

Figura 4. Representación de la placa de cultivos 24 well donde se realizo el experimento. Se sembraron células en las filas B, C y D. Y los pocillos de las columnas 1, 2 y 3 son las células control, mientras que los pocillos de las columnas 4, 5 y 6 son los pocillos con tratamiento antimitótico.
Los resultados obtenidos de procesar las fotos con la red neuronal creada, son los núcleos contados en las 16 fotos de cada uno de los pocillos de la placa de cultivo. En la siguiente tabla comparamos los resultados obtenidos tras procesar las imágenes con la red neuronal de IA realizando un entrenamiento semiautomatico con el software Cellsens (columna 3, rojo), con los resultados obtenidos con un proceso de segmentación con post-procesado watershed (columna 1) y con post-procesado sin watershed (columna 2). Las celdillas en verde son los núcleos contados en las 16 fotos de cada uno de los pocillos con tratamiento antimitótico.

Como podemos ver en la primera columna (post-procesado watershed) tenemos más núcleos y por tanto más células que en la segunda y en la tercera columnas, para todos y cada uno de los pocillos sean o no con tratamiento. Esto es debido a que separa los núcleos de células diferentes que están adyacentes (lo cual no pasa en la segunda columna) y además los núcleos raros los separa en varios núcleos como vimos en la figura 2B abajo.
En la segunda columna (post-procesado sin watershed) tenemos por lo general menos núcleos y por tanto menos células que en las otras dos columnas, puesto que comete el error de contar como un solo núcleo aquellos que son adyacentes, sin embargo los núcleos raros los cuenta bien mayoritariamente, puesto que no comete el error de separar en varios núcleos aquellos de formas extrañas.
Por último, en la tercera columna (IA software Cellsense) los núcleos adyacentes los cuenta como separados, además este tipo de núcleos son especialmente abundantes en las los fotos de los pocillos con alta confluencia de células (pocillos sin tratamiento donde las células se dividen constantemente). En esta tercera columna los núcleos raros de las fotos de los pocillos con tratamiento antimitótico los identifica perfectamente sin cometer el erro de separarlos en varios núcleos. De manera que utilizando el procesamiento de las fotos con la red neuronal creada en Cellsens de manera semiautomática (columna 3) podemos asegurar que son identificados todos los núcleos, tanto normales como raros y además separa los núcleos adyacentes de los pocillos con alta confluencia celular.
A partir de aquí, utilizando el procesamiento con la red neuronal de IA podemos seguir analizando la placa de cultivo y determinando el área media de las células, el número de núcleos raros por pocillo, los diferentes marcajes y sus intensidades tanto citosólicos como nucleares y muchos más parámetros de interés para el investigador mediante el programa ScanR de HCA.
AUTOR
José Ángel Rodríguez Alfaro.
Responsable del Servicio Microscopía y Análisis de Imagen.
Hospital Nacional de Parapléjicos.
Toledo, España.