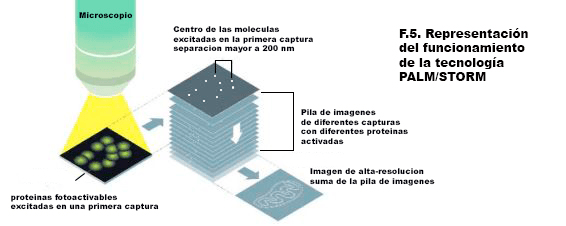
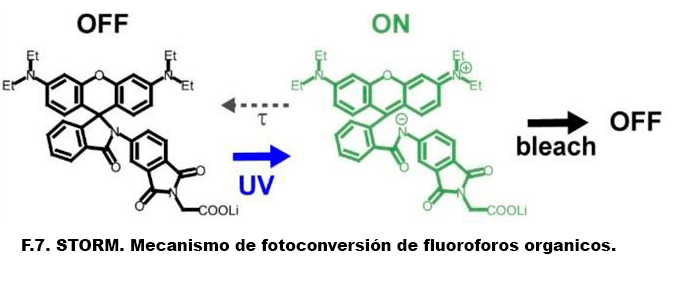
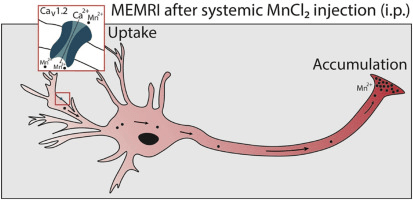
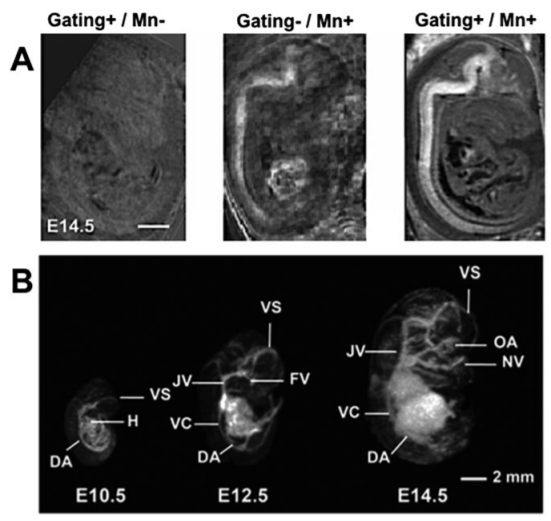
En esta nueva entrada, dejamos a un lado las técnicas para hablar de seguridad y funcionamiento en un laboratorio de Resonancia Magnética de alto campo.
A) Información de seguridad
El acceso a la sala tiene que estar regido por unas normas de funcionamiento tanto para la entrada, operación y emergencia en caso de desactivación del imán. Todo el personal que entre tiene que estar informado de estas normas y ser consciente que va a entrar en una zona con altos campos magnéticos la cual entraña un riesgo importante.
Estas normas tienen que aplicarse a:
1) Imán, seguridad y delimitación de la zona
El campo magnético es de 7T, las líneas de fuerza del campo magnético (línea de 5 Gauss) se van a distribuir en un volumen tridimensional alrededor del imán.
Es conveniente señalar o indicar sobre el suelo la distancia que cubre este volumen, de esta manera nos aseguraremos la zona donde se tienen que extremar las precauciones. Esta delimitación se encuentra a metro y medio del imán.
Concretamente, en nuestro laboratorio la Linea de 5 Gauss se encuentra en toda la sala del equipo.
Área de acceso controlada
Solo podrá entrar personal autorizado que haya sido previamente informado de los riesgos.
– Está prohibida la entrada a todas aquellas personas que lleven implantes metálicos ferromagnéticos, marcapasos o piercings (que no puedan quitarse).
– Antes de entrar en la sala de RM habrá que depositar los teléfonos móviles, relojes, tarjetas con banda magnética, abonos transporte, llaves, monedas, clips, tijeras, pinzas, imperdibles de las etiquetas de las cubetas (todos esos objetos se quedan en los bolsillos y es muy frecuente que se nos olvide depositarlos en el sitio adecuado). En general todos aquellos objetos que sean atraídos por el imán y que puedan actuar de proyectil.
2) Instalación de señales de precaución y advertencia
En la entrada a la sala del imán se colocarán señales informativas de los riesgos y precauciones que supone trabajar con campos magnéticos altos. Estos carteles son suministrados por Bruker y en ellos se informa de la entrada a una zona con un alto campo magnético y con alto nivel de ruido, la entrada restringida a determinadas personas, y objetos que no se pueden introducir.
3) Plan de emergencia
– Dispositivos físicos de emergencia:
En la sala de control, al lado de la consola existirá un pulsador de emergencia ON/OFF, este dispositivo apagará toda la parte electrónica, sin desactivar el imán, por supuesto.
– Para la desactivación del imán, se colocará un pulsador ROJO señalado como “quench”. Su localización en la sala de RM deber ser conocida y de fácil acceso, deberá ser pulsado en caso de accidente que implique peligro de vida. Este pulsador, provoca la desactivación del imán, extrayendo el helio líquido liquido por la chimenea. Una vez pulsado, se saldrá lo más rápido posible de la sala.
– Como medida de seguridad adicional sería deseable instalar unos detectores de concentración de oxígeno. Si el helio por algún motivo, se pudiese evaporar, su volumen aumentaría 760 veces. Esto produciría un aumento de la presión dentro del criostato obligando al helio a salir por la válvula de seguridad. El helio es un gas inodoro, incoloro e insípido y menos denso que el aire. En el caso de que se produjese la evaporación, éste ascendería al techo de la sala, reemplazaría al aire y al respirar, produciría asfixia y congelación, de ahí que sea recomendable instalar el detector de concentración de oxígeno.
4) Instrucciones para el personal de seguridad y de limpieza
La entrada a la sala está prohibida a menos que sean supervisados por el operador de RM y advertirles de los riesgos que conlleva trabajar con altos campos magnéticos. La limpieza de la sala se hará con material que haya sido supervisado previamente por el operador de RM, estando localizado en la misma sala. Para evitar posibles accidentes, el acceso estará prohibido a este personal.
5) Primeros auxilios y antiincendios
Como todo laboratorio se dispondrá de un extintor contraincendios, uno para sala técnica y un extintor no magnético para la sala del imán.
6) Transporte y almacenaje de líquidos criogénicos
Esta última tecnología de imanes requiere un mantenimiento mínimo de llenado de líquidos criogénicos (Helio líquido). El llenado de Helio será hecho por personal especializado.
7) Monitorización y ventilación del aire en la sala del imán
Es muy importante mantener una buena climatización de la sala, sobretodo en la sala técnica, el aire acondicionado debe mantener la temperatura en torno a los 21ºC tanto en verano como en invierno.
8) Condiciones ambientales de ruido.
Para evitar el exceso de ruido provocado por los gradientes del imán durante la realización de los experimentos, la puerta entre la sala de control y la del imán ha de permanecer cerrada. Un ruido adicional y que siempre es constante es el provocado por la toda la electrónica del equipo, por tanto, la puerta de la sala técnica tiene que estar cerrada en todo momento.
Todas estas normas son de aplicación general para cualquier instalación de Resonancia Magnética tanto para la clínica como para la experimental.
Adicionalmente existe una regulación de seguridad tanto por país, como una regulación especifica para trabajar en áreas con altos campos
B) Aspectos básicos de seguridad
1) Leer las instrucciones de seguridad: todos los usuarios deben conocer el manual de seguridad y el plan de emergencia al trabajar con sistemas de Resonancia Magnética.
2) Tener localizado el botón de emergencia on/off situado al lado de la consola.
3) Tener localizado el pulsador rojo para la desactivación del imán (Botón del Quench).
4) Toda persona que entre en la sala debe de tener conocimiento del modo de trabajo en la sala de RM y haber sido instruido (incluido el personal de seguridad y el de limpieza, si acceden a la sala) por la persona responsable del equipo de RM.
5) El usuario además, debe saber la localización del botiquín, extintores, extintores no magnéticos y de todos los dispositivos que tengan que ser utilizados en caso de emergencia.
C) Procedimiento general ante una emergencia
Todos los usuarios y el personal que entre en la sala debe ser capaz de proceder en caso de emergencia. Es decir, deberá saber que:
1) En todas las circunstancias, hay que informar a los miembros responsables de la instalación.
2) En caso de fuego o de descarga eléctrica. Desconectar el sistema inmediatamente de la red principal por medio del interruptor de emergencia on/off.
3) Si un objeto ha sido atraído por el imán con riesgo de peligro de vida, pulsar inmediatamente el botón de emergencia de quench para desactivar el imán.
4) Si un objeto ha sido atraído por el imán sin riesgo de peligro de vida, no intentar quitarlo. En este caso habrá que avisar al técnico de la casa comercial.
5) Abandonar la sala inmediatamente.
D) Procedimiento en caso de fuego
Recordad que ante cualquier caso de emergencia, siempre habrá que avisar al responsable.
– Si hay fuego en la sala técnica:
Todos los componentes serán apagados con el interruptor de emergencia ON/OFF de la consola o desconectarlo desde cada uno de los sistemas principales. Cada modulo tiene su propio sistema de apagado.
En principio, en la sala técnica si se pueden utilizar los extintores convencionales tomando las respectivas medidas de seguridad. Aún así, sería recomendable tener extintores no magnéticos en las proximidades del equipo de RM.
– Si hay fuego en el interior de la sala del imán:
En este caso, el sistema eléctrico del imán no tiene porqué apagarse inevitablemente. La decisión de apagarlo dependerá del riesgo que ello implique, se utilizará el botón ON/OFF situado al lado de la consola.
SOLO se pueden utilizar extintores no magnéticos en la zona del imán y estos tienen que estar debidamente marcados como no magnéticos y conocer su localización.
Mientras que el imán esté activo solo se podrá utilizar material no magnético. Si hay que avisar a los bomberos, tener especial cuidado con las máscaras de gas y los equipos de respiración ya que suponen un riesgo de vida. En el caso de que tengan que entrar estos dispositivos, es obligatorio que el imán sea desactivado y que el campo magnético se anule por completo antes de entrar.
E) Procedimiento en caso de una emergencia médica
Si fuese necesaria asistencia médica, ésta deberá producirse fuera de la sala del imán. Si no fuese posible atender a la persona fuera, habrá que desactivar el imán antes de entrar con cualquier equipamiento magnético.