En una de nuestras primeras newsletters, os hablábamos sobre la metodología SWATH-MS (Sequential Window Acquisition of All Theoretical Fragment Ion Mass Spectra). Este tipo de tecnología, de adquisición independiente de datos (DIA) de espectros en espectrometría de masas, en estos años se ha revelado como una de las más prometedoras para mejorar la cobertura y la cuantificación de proteínas en mezclas complejas.
En esta newsletter vamos a profundizar un poco más sobre las estrategias y aplicaciones de este enfoque en análisis “ómicos”
¿Qué es una adquisición independiente de datos de espectros (DIA)?
El propio nombre lo indica. Es un método de adquisición con el que el espectrómetro nos va a recoger todos los datos de espectros de la muestra. Para un método de adquisición dependiente de datos el espectrómetro va a seleccionar únicamente aquellos iones que cumplan con unos criterios previos establecidos, mientras que en los métodos DIA, el espectrómetro va analizando por ventanas todos los iones que encuentre (Figura 1).
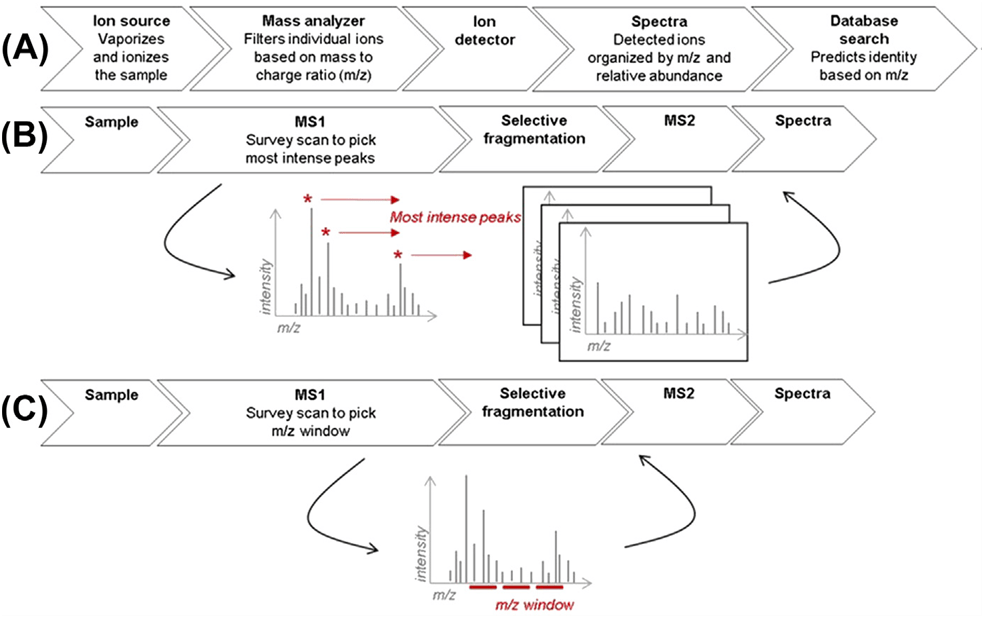
Figura 1: Espectrometría de masas en tándem para proteómica cuantitativa. (A) Esquema básico de espectrometría de masas. (B) Adquisición dependiente de datos. Los iones precursores se seleccionan en función de un umbral predefinido. (C) Adquisición independiente de datos. Los iones precursores se seleccionan en una ventana m/z.
¿Por qué SWATH-MS?
SWATH-MS es como ha denominado Sciex a los métodos DIA de sus equiposzenoTOF, QTOF y TripleTOF de escaneo rápido, en los que se adquiere en ventanas de forma secuencial todos los fragmentos de masas de los iones que se registran previamente. Estas “ventanas” son intervalos de m/z que pueden ser de un ancho fijo o variables.
La ventaja que presenta tomar ventanas de tamaño variable es que podemos seleccionar intervalos más estrechos en zonas de nuestra cromatografía donde haya una mayor densidad de los precursores de analitos. Así, se logra una mejor especificidad en matrices complejas, ecualizando la densidad de precursores en cada una de las ventanas de aislamiento (Figura 2).

Figura 2: Investigación de anchos de ventana variables del Q1 para la adquisición de SWATH. Los histogramas de densidad m/z construidos a partir de los datos TOF MS para la muestra de interés (línea azul) se pueden usar para construir un patrón de ventana de tamaño variable (línea roja) usando la calculadora de ventana variable SWATH.3 En la figura se muestra una distribución m/z típica y un patrón de ventana Q1 utilizado para muestras de proteoma digeridas.
Cada casa comercial ha intentado implementar este tipo de adquisición en sus instrumentos con rendimientos más o menos limitadospara la interpretación de matrices complejas. Para conseguir explotar al máximo los datos obtenidos, se necesitan herramientas informáticas que nos ayuden en el análisis.
El desafío analítico que presenta la interpretación de datos DIA es la distinción de múltiples péptidos que están coaislados y cofragmentados en la misma ventana de selección de precursores. Es decir, perdemos esa relación directa que teníamos en ensayos DDA del precursor con sus iones fragmentos.
Estrategias de análisis
Para la interpretación de datos DIA podemos ayudarnos de software basados en:
- Utilización de bibliotecas espectrales.
- Sin bibliotecas espectrales.
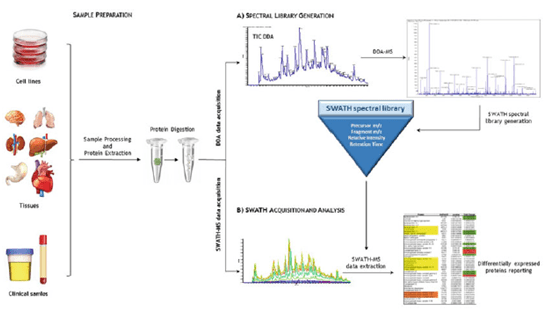
Figura 3: Representación esquemática de las estrategias para construir la biblioteca espectral en el análisis SWATH-MS. (A) La biblioteca espectral se obtiene de un experimento DDA simple. Contiene toda la información sobre un péptido identificado determinado (como tR, precursor m/z y espectros MS/MS). (B) Una vez realizada la biblioteca, las muestras se ejecutan en modo SWATH
- Utilización de bibliotecas espectrales.
De forma sencilla, una biblioteca espectral generalmente se compone de un conjunto de coordenadas que incluyen el precursor m/z, el ion fragmentado m/z, la intensidad relativa de los iones fragmentados y el tiempo de retención estandarizado (RT) para cada precursor peptídico y sus fragmentos. La biblioteca espectral se utiliza para extraer las cromatografías iónicas de los péptidos objetivos de los espectros DIA.
El éxito de los experimentos DIA depende de la calidad de las bibliotecas espectrales utilizadas para la búsqueda en la base de datos. Con frecuencia, estas bibliotecas deben generarse mediante experimentos de adquisición dependiente de datos (DDA) fraccionados de las mismas muestras con las que se va a realizar el ensayo DIA. Esto que requieren mucho tiempo y trabajo.
¿Cómo construimos una biblioteca espectral? En teoría, todas las herramientas de software de búsqueda de espectro MS/MS pueden usarse potencialmente para construir las bibliotecas DIA.
Podemos resumirlo en tres pasos: i) recopilación de espectros de péptidos identificados con confianza en los motores de búsqueda, ii) integración de espectros del mismo ion peptídico en un único espectro representativo y tiempo de retención normalizado, iii) control de calidad para eliminar espectros no confiables Recientemente se han publicado varios algoritmos que permiten generar bibliotecas teóricas mediante una predicción eficaz del tiempo de retención y la intensidad de los iones fragmento.
Para generar bibliotecas teóricas fiables que puedan utilizarse en experimentos SWATH, se han desarrolladoherramientas de aprendizaje profundo para el análisis SWATH, con el fin de mejorar la sensibilidad y especificidad de los datos generados por espectrómetros de masas Q-TOF.
- Sin bibliotecas espectrales.
En principio, si un péptido no está presente en la biblioteca espectral, no podrá analizarse utilizando métodos basados en bibliotecas. Así, se han desarrollado alternativas a los análisis DIA denominados “libres de bibliotecas” o método centrado en el espectro.
Aunque la búsqueda sin bibliotecas centrada en el espectro ha demostrado su potencial para identificar péptidos nuevos, la mayoría de los estudios publicados todavía utilizan el enfoque de extracción dirigida basado en bibliotecas espectrales.
Aplicaciones.
Bien, después de este despliegue de información sobre qué es un análisis DIA y sus estrategias de análisis, ¿en qué aplicaciones según las necesidades de mi trabajo de investigación puedo considerarlo?
Si el objetivo de nuestro proyecto es identificar el número máximo de proteínas a partir de un número limitado de muestras de una determinada complejidad, DIA no debería ser nuestro método de elección. Para este tipo de estudios la metodología basada en fraccionamiento de péptidos combinada con gradientes largos y métodos DDA tienen una mayor profundidad en identificaciones.Aunque tengamos una limitación con la cantidad de muestra necesaria para la realización del ensayo.
En los enfoques DIA, las concentraciones requeridas son menores (0.1-1µg) y la principal ventaja es poder utilizar gradientes de cromatografía cortos para poder analizar un proteoma tisular completo
DIA-MS está diseñado para combinar el potencial de la metodología no dirigida (discoveryshotgun, DDA) y el enfoque dirigido (SRM/MRM).En concreto, SWATH-MS se presentó como una alternativa para superar las limitaciones de métodos de cuantificación sin etiqueta. Al tratarse de un método insesgado, capaz de cuantificar un gran número de péptidos con una consistencia y precisión similares a las de métodos dirigidos como MRM, lo que convierte en una estrategia prometedora para el descubrimiento de biomarcadores a partir de cribados a gran escala.
Cuando se evalúa la opción de enfoques DIA, no solo debe considerar la identificación objetiva de proteínas y la precisión cuantitativa, que se han realizado para los datos SWATH, sino que también debe tener en cuenta otros factores cruciales, incluido el objetivo de la investigación, la estrategia de adquisición de datos DIA, rendimiento, consumo de cantidad de muestra, instrumentos de MS, viabilidad y costo del análisis.
Referencias aplicaciones
Klont F, Jahn S, Grivet C, König S, Bonner R, Hopfgartner G. SWATH data independent acquisition mass spectrometry for screening of xenobiotics in biological fluids: Opportunities and challenges for data processing. Talanta. 2020 May1;211:120747. doi: 10.1016/j.talanta.2020.120747. Epub 2020 Jan 15. PMID: 32070597.
Anjo, S.I.; Santa, C.; Manadas, B. SWATH-MS as a Tool for Biomarker Discovery: From Basic Research to Clinical Applications.Proteomics2017,17doi.org/10.1002/pmic.201600278
Finamore F, Cecchettini A, Ceccherini E, Signore G, Ferro F, Rocchiccioli S, Baldini C. Characterization of Extracellular Vesicle Cargo in Sjögren’s Syndrome through a SWATH-MS Proteomics Approach. International Journal of Molecular Sciences. 2021; 22(9):4864. https://www.mdpi.com/1422-0067/22/9/4864
Hallal S, Azimi A, Wei H, Ho N, Lee MYT, Sim H-W, Sy J, Shivalingam B, Buckland ME, Alexander-Kaufman KL. A Comprehensive Proteomic SWATH-MS Workflow for Profiling Blood Extracellular Vesicles: A New Avenue for Glioma Tumour Surveillance.International Journal of Molecular Sciences. 2020; 21(13):4754. https://www.mdpi.com/1422-0067/21/13/4754
Turner N, Abeysinghe P, Flay H, Meier S, Sadowski P, Mitchell MD. SWATH-MS Analysis of Blood Plasma and Circulating Small Extracellular Vesicles Enables Detection of Putative Protein Biomarkers of Fertility in Young and Aged Dairy Cows Journal of Proteome Research 2023 doi.org/10.1021/acs.jproteome.3c00406
AUTOR
Gemma Barroso García, MSc.
Responsble SAI-Proteómica.
Hospital Nacional de Parapléjicos.
Toledo, España.
Para más información, dudas o solicitud de presupuestos puedes contactarnos en: unidadproteomica.hnp@sescam.jccm.es
Si quieres conocer más sobre nosotros: https://hnparaplejicos.sanidad.castillalamancha.es/profesionales/investigacion/servicios-apoyo/proteomica
Síguenos en:
www.linkedin.com/in/servicios-de-apoyo-a-la-investigación-sais-hnp
