Los últimos avances en citometría de flujo están permitiendo a los investigadores llevar a cabo experimentos multiparamétricos de gran complejidad con paneles de más de 40 colores. Para conseguir la mayor precisión y reproducibilidad de los datos obtenidos en citometría de flujo existen diferentes consideraciones que se deben tener en cuenta a la hora de diseñar un panel multicolor con éxito.
- CONOCER EL EQUIPAMIENTO
Un análisis multiparamétrico implica la utilización de diferentes fluoróforos o fluorocromos, cuya medida viene dada por la excitación de los mismos con diferentes láseres, y por la medida de su emisión de fluorescencia en detectores concretos. Por tanto, conocer la configuración de tu equipo es crucial para saber qué fluorocromos pueden ser medidos en él, y con qué detectores.
- FILTROS
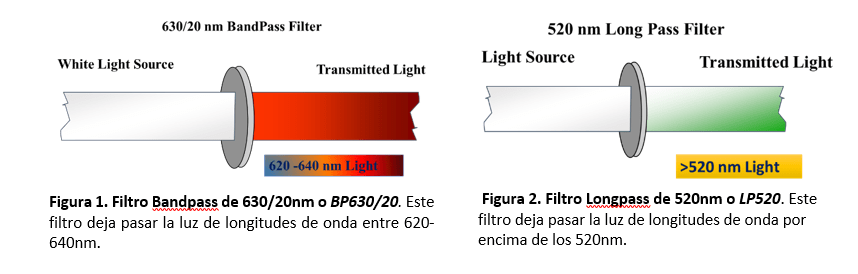
Son los que ayudan a dirigir la dispersión de luz y/o la fluorescencia emitida hacia detectores concretos. Existen dos tipos de filtro. Por un lado, los filtros de paso largo o Longpass (LP, Fig.1), que solo dejan pasar la luz de longitudes superiores a las de longitudes indicadas en el filtro. Y los filtros de banda o Bandpass (BP, Fig.2), los cuales sólo dejan pasar luz de un determinado intervalo de longitud de onda.
- DETECTORES
Los detectores son los componentes del citómetro que van a convertir la señal luminosa en una señal eléctrica, es decir, gracias a la absorción de fotones van a emitir electrones. Hay 2 tipos principales: detector de FSC (dispersión frontal), que es un fotodiodo y los detectores de SSC (dispersión ortogonal) y fluorescencia, que son los fotomultiplicadores. En los citómetros más nuevos como el CytoFLEX (Beckman Coulter) se utilizan fotodiodos de avalancha, que al ser más sensibles permiten una mayor resolución.
- LÁSER
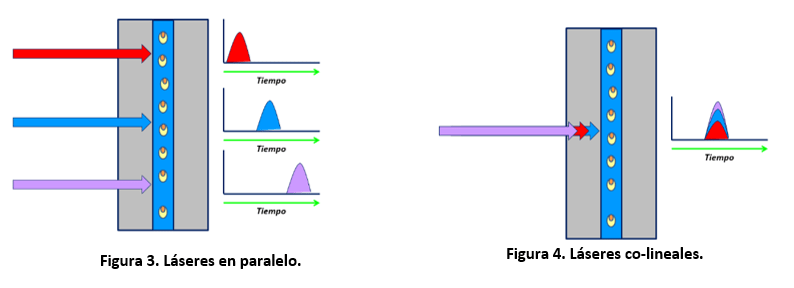
Los citómetros puede tener los láseres en paralelo o ser co-lineales. Los láseres en paralelo tienen una separación espacial o “laser delay” entre los puntos de incidencia de los láseres en el punto de interrogación, de forma que primero incide un láser, después el láser de referencia (que suele ser el 488 nm), y así con los demás láseres sucesivamente (ver figura 3). Así, las señales excitadas por cada uno de los láseres se recogen a tiempos distintos, con lo que se pueden utilizar juntos en el mismo panel, fluorocromos que se exciten con varios láseres. Sin embargo, en los citómetros co-lineales, no existe esta separación espacial entre las líneas de láser (figura 4), con lo que hay una mayor limitación de fluoróforos que se pueden utilizar, ya que existen fluoróforos que se pueden excitar con diferentes líneas de láser, pero que se recogen en el mismo detector.
En función de la configuración de láseres, filtros y detectores que tenga cada modelo de citómetro, vamos a tener que diseñar nuestro panel con fluoróforos que puedan ser “leídos” por el equipamiento disponible.
En nuestro caso, el SAI Citometría de Flujo cuenta con 2 citómetros analizadores y 1 citómetro separador: BD FACS Canto II, citómetro analizador equipado con 3 Láseres (405 nm-488 nm-633 nm), que permite la detección de 8 colores; CytoFLEX S (Beckman Coulter), equipado con 4 láseres (405 nm-488 nm-561 nm-638 nm) que permite la detección de 13 colores, gracias a la actualización que hemos podido realizar mediante la obtención de una subvención en la convocatoria de infraestructuras del ISCIII de 2021; citómetro separador BD FACS ARIA IIu, 3 láseres (405 nm-488 nm-633 nm), que permite la detección de 9 colores. Todos los citómetros del CITF-SAI-HNP tienen los láseres en paralelo. Pinchando en el nombre de cada equipo accederéis a la configuración de cada uno de ellos donde encontraréis el conjunto de detectores y filtros de los cuales están compuestos: FACS Canto II, CytoFLEX S y FACS Aria II.
- SELECCIÓN DE ANTICUERPOS
En el caso de utilizar anticuerpos para caracterizar las poblaciones celulares que tenemos en una muestra (por ejemplo, caracterización de las poblaciones presentes en una médula espinal lesionada), es importante tener 2 cosas en cuenta:
- MARCADORES: decidir qué marcadores son los que definen la población celular de interés
- CLONES: determinar si hay clones descritos con mejor reactividad/mayor afinidad que otros en un determinado tejido/panel. Tened en cuenta que algunos clones no funcionan bien cuando las células son sometidas a un tratamiento.
- CLASIFICACIÓN DE ANTÍGENOS
En Citometría de flujo el antígeno es la molécula en una célula que intentamos identificar.
Los antígenos se clasifican de acuerdo a su densidad de expresión, es decir, a la abundancia relativa del Ag en la célula de interés, la cual nos ayudará en la elección del fluorocromo adecuado.
Podemos definir los antígenos según la población que nos permitan definir:
- Prioritarios: Son aquellos absolutamente necesarios para definir los linajes principales presentes en nuestra muestra. Ej: GFAP como específico de astrocitos o CD3 como marcador específico de linfocitos T.
- Secundarios: Nos ayudan a definir subpoblaciones. Ej: dentro de los linfocitos T (CD3+) podemos definir linf T helper por la expresión de CD4.
- Desconocidos: Son nuestra hipótesis, aquellas moléculas que queremos estudiar.
- De lujo/bonus: Antígenos no necesarios que nos ayudan a refinar nuestro análisis.
Por otro lado, se pueden clasificar de acuerdo al modo de expresión:
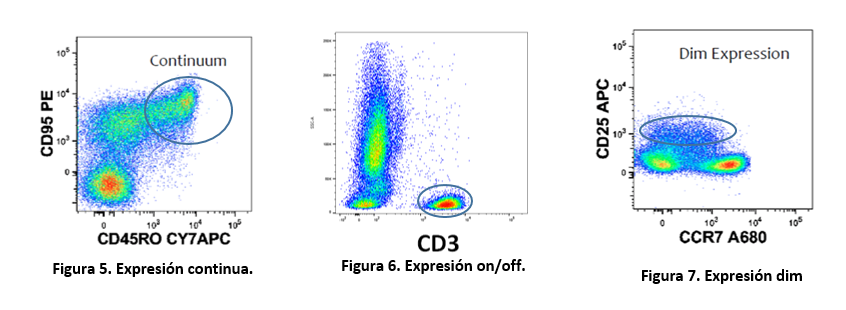
• Continua: dentro de la población existen células con diferente nivel de expresión, de forma que el patrón de marcaje es continuo (Fig.5), no permitiendo definir fácilmente cuál es la población positiva (necesarios controles “Fluorescence Minus One” para el análisis)
• Positivo/negativo = on/off: la población positiva expresa el marcador a una alta densidad y de forma homogénea, con lo que se pueden distinguir perfectamente del resto de poblaciones celulares (Fig.6).
• dim (tenue): expresión muy débil del antígeno en cuestión.
Es muy importante conocer los antígenos o marcadores específicos de las células que queremos estudiar. Para ello, se pueden utilizar diferentes fuentes bibliográficas que permiten conocer dicho marcador, qué fluorocromos disponibles existen y el espectro de emisión de cada uno de ellos: https://www.biolegend.com/essential_markers; https://www.thermofisher.com/es/es/home/life-science/cell-analysis/labeling-chemistry/fluorescence-spectraviewer.html.
- SELECCIÓN DE FLUOROCROMOS
Una vez que sabemos qué fluorocromos podemos detectar en nuestro equipamiento, y que hemos categorizado los antígenos/marcadores en función de su expresión y la población que definen, tenemos que emparejar los marcadores con los fluorocromos, y para ello tenemos que tener en cuenta el brillo de los mismos.
- MÁS BRILLANTES O BRIGHT FLUOROCHROMES se deben usar para antígenos de expresión desconocida (hipótesis), alguna población celular muy poco representada (eventos raros) o para antígenos de baja expresión (dim/tenue).
- MENOS BRILLANTES O DIM FLUOROCHROMES se emplean marcadores con altos niveles de expresión, poblaciones muy representadas, marcadores que definen linajes con expresión de tipo on/off.
Cuando diseñamos un panel, si utilizamos un fluorocromo con menos brillo (fig. 8 panel izq, marcador CD25-FITC) para un antígeno que se expresa de forma tenue (dim), puede que no seamos capaces de distinguir adecuadamente la población del fondo/población negativa. En cambio, podemos usarlo para distinguir una población de linaje principal o secundario con expresión de tipo on/off, y utilizar el más brillante para analizar el antígeno tenue (Fig.8 panel dcho marcador CD25-PE)

- SPILLOVER Y CO-EXPRESIÓN: el spillover se define como el solapamiento de la fluorescencia emitida por un fluorocromo en un canal secundario debido a la física de la fluorescencia, y que tiene que ver con el espectro de emisión de un fluorocromo. Cuando tenemos marcadores que se co-expresan, debemos elegir fluorocromos con el mínimo spillover el uno con el otro, es decir, que el solapamiento de fluorescencia sea el menor.
- AUTOFLUORESCENCIA: algunos tipos celulares tienen una gran autofluorescencia basal (típicamente emisión en torno a 530 nm excitada por los láseres 405 nm y 488 nm). En este caso, para el análisis de marcadores tenues o los que son nuestra hipótesis, se debe elegir un fluorocromo que no se detecte en los detectores en los que se puede recoger más fácilmente la autofluorescencia.
Existen páginas web comerciales donde se puede consultar el índice de brillo de cada de cada fluorocromo: Fluorophore Brightness Index, así como visores de espectros que nos pueden ayudar a elegir los fluoróforos con menor solapamiento de fluorescencia (https://www.thermofisher.com/es/es/home/life-science/cell-analysis/labeling-chemistry/fluorescence-spectraviewer.html )
Además, debemos tener en cuenta que las células muertas pueden unir anticuerpos inespecíficamente, por lo que añadir un tinte de viabilidad celular que nos permita eliminar las células muertas del análisis es crítico.
CONCLUSIÓN
Aunque a primera visto os puede parecer misión imposible el diseño de paneles multiparamétricos, no debemos olvidar que existen multitud de páginas que permiten su automatización, incluyendo las principales casas comerciales como: Fluorofinder (https://www.fluorofinder.com) o Chromocyte (https://www.chromocyte.com/calculate).
Además, el SAI Citometría está a vuestra entera disposición, si necesitáis ayuda para el diseño de un panel o dudáis con la elección de algún fluoróforo no dudéis en contactar con nosotras. Os animamos a poner en práctica vuestras destrezas en citometría con el diseño del mayor panel realizado hasta la fecha en el SAI con la ampliación del Cytoflex S a 13 colores.
AUTOR
Ángela Marquina Rodríguez
Técnico Titulado Superior del Servicio de Citometría.
Hospital Nacional de Parapléjicos.
Toledo, España.
Referencias
- FlowPost-Its “Panel design” Mayo 2022 Flow Cytometry Core Facility at Memorial Sloan Kettering Cancer Center (MSKCC).
- “Curso teórico-práctico de Citometría de flujo 2022 (3ª edición)”. Junio 2022. Servicio de Citometría de Flujo del Hospital Nacional de Parapléjicos (CITF-SAI-HNP).
