En la anterior newsletter, hicimos una breve introducción a las diferentes opciones de experimentos en proteómica cuantitativa y la importancia de la identificación y cuantificación integral de todas las proteínas en un sistema biológico, para revelar las funciones de las proteínas en los procesos biológicos, fisiológicos y patológicos. En esta nueva entrega nos enfocamos en los métodos basados en marcaje o proteómica cuantitativa multiplexada, con etiquetado o marcaje químico. Estos métodos isotópicos se caracterizan por una mejor reproducibilidad y precisión comparado con los métodos “label-free”. Otra de sus principales ventajas es el multiplexado, que permite analizar en paralelo varias muestras, lo que ahorra tiempo de análisis y aumenta la fiabilidad general del método.
Las modificaciones químicas de los péptidos se obtienen por hidrólisis enzimática de las proteínas. Los reactivos utilizados están diseñados para unirse a los grupos amino, sulfhidrilo o carboxilo de los péptidos. Los isótopos pesados que incorporan estos reactivos son 13C, 15N, 18O y 2H. La incorporación de los marcajes hace que se formen péptidos que poseen las mismas propiedades químicas y cromatográficas, pero con diferente masa molecular para poder distinguirlos en el espectro de MS, permitiendo así la comparación entre péptidos marcados (Heavy) y sin marcar (Light). El deuterio es el menos utilizado ya que existe riesgo de variación de los tiempos de retención en LC de un mismo péptido cuando tiene diferente número de modificaciones.
Los métodos de marcaje isotópico podemos clasificarlos según diferentes criterios (Tabla 1).
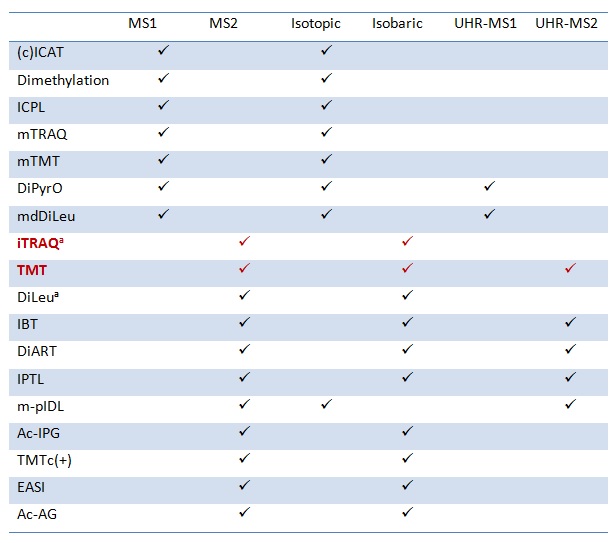
Tabla 1. Clasificación de los enfoques de marcaje químico basados en diferentes criterios
a Aunque las etiquetas de las series iTRAQ y DiLeu tienen diferencias de mDa en las masas de precursores, se clasifican como isobáricas ya que las diferencias de mDa no se revelan a nivel de MS1 en el uso general y ya se las denomina habitualmente isobáricas.
Para intentar simplificarlo y que no os resulte tedioso, describiremos las aproximaciones como cuantificaciones basadas en MS1 y MS2 (Figura 1) centrándonos, en esta ocasión, exclusivamente en los ensayos basados en MS1.
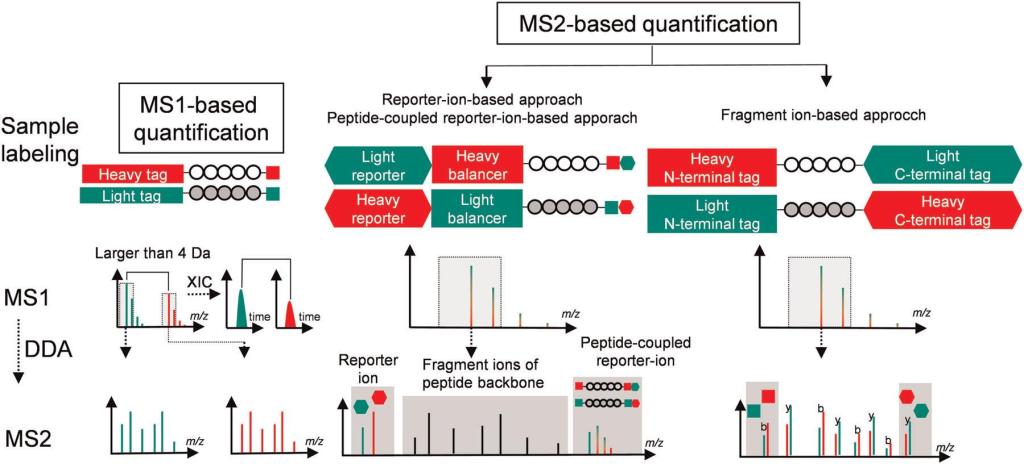
Figura 1. Descripción esquemática de la cuantificación basada en MS1 y MS2. (Xiaobo Tian et al., Mass Spectrom Rev. 2021;1–31)
Antes de discutir estrategias de etiquetado específicas, os refresco las características principales de los dos modos principales de adquisición de espectros de MS2, a saber, adquisición dependiente de datos (DDA) y adquisición independiente de datos (DIA), ya que están estrechamente vinculadas a las ventajas y desventajas de los enfoques de cuantificación multiplexados.
En la mayoría de los métodos multiplexados, la recopilación de espectros de MS2 se realiza en modo DDA. Este método de adquisición tiene un sesgo hacia las especies de mayor abundancia y limita la detección de péptidos de baja abundancia y, en última instancia, proteínas (Figura 2). El número de picos que se pueden aislar para la fragmentación aumenta con la velocidad de exploración del espectrómetro de masas, pero generalmente hay más picos en los espectros MS1 de los que el instrumento puede aislar en el período de tiempo del análisis, dando como resultado que se ignoren los picos menores. Este problema aumenta con la complejidad de las muestras lo que lleva al hecho de que las muestras muy complejas a menudo deben fraccionarse antes del análisis LC-MS final.
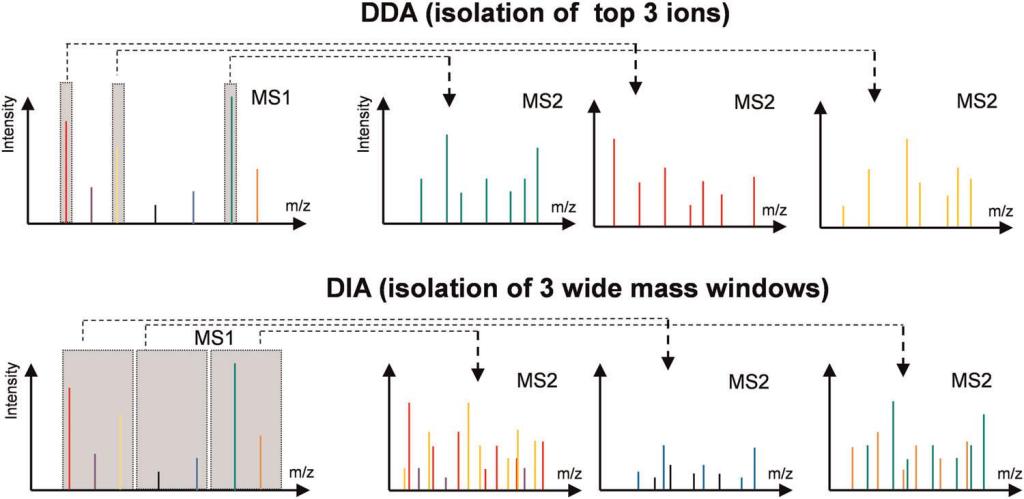
Figura 2. Diferencia en el aislamiento en MS1 para los modos DDA y DIA
A pesar de estas limitaciones, la DDA sigue siendo la estrategia de adquisición de datos más utilizada en la proteómica de descubrimiento actual, debido a los flujos de trabajo de adquisición y procesamiento de datos bien desarrollados que se pueden implementar en casi cualquier instrumento de EM.
En contraste con DDA, el aislamiento de precursores para la fragmentación en DIA no se basa en las intensidades máximas en MS1. En cambio, todos los precursores presentes dentro de una ventana de aislamiento predefinida serán fragmentados y analizados juntos, como se muestra en la Figura 2. DIA aborda en gran medida el problema de los valores perdidos de DDA, sin embargo, se pierde el vínculo directo entre iones precursores y fragmentos.
Los datos de MS1 como de MS2 se pueden utilizar para la cuantificación en modo DIA. Si bien la aplicación del etiquetado de isótopos estables se usa ampliamente en el modo DDA, todavía está en sus inicios cuando se trata de DIA.
CUANTIFICACIÓN BASADA EN MS1
Los enfoques cuantitativos basados en MS1 consisten en la adición de distintas masas a los péptidos mediante marcaje isotópico, de modo que los mismos péptidos derivados de diferentes muestras tengan masas diferentes. La cuantificación se logra comparando las áreas o intensidades de los picos de iones peptídicos para cada marcaje a nivel de MS1. Posteriormente, la información cuantitativa a nivel de péptido debe transferirse al nivel de proteína, esta estrategia es común para DDA y DIA. Por lo general, se incorpora un desplazamiento de masa de al menos 4 Da en el marcaje para evitar la superposición entre las envolventes de isótopos de muestras Heavy y Ligth. La superposición resultante de cambios de masa más pequeños (<4 Da) hace que la cuantificación relativa sea más difícil ya que se requiere una etapa adicional de deconvolución.
Los distintos isótopos para la cuantificación basada en MS1 se pueden incorporar a las muestras mediante tres enfoques:
(1) Etiquetado químico (p. Ej., ICAT y mTMT) con etiquetas sintéticas dirigidas a grupos reactivos (amina o tiol).
(2) Marcaje enzimático, donde las proteínas se digieren enzimáticamente en presencia de agua marcada con 18O, incorporándose dos átomos de 18O a los grupos carboxilo C-terminales generados en el proceso de digestión.
ICAT
ICAT fue el primer marcador isotópico, consta de tres partes funcionales como se muestran en la figura 3:
(1) Un resto de biotina;
(2) Un enlazador isotópico, que contiene ocho átomos de 2H o 1H para marcar diferencialmente péptidos de diferentes muestras;
(3) Un grupo yodoacetamida, que reacciona específicamente con el grupo sulfhidrilo en la cadena lateral de los residuos de cisteína.
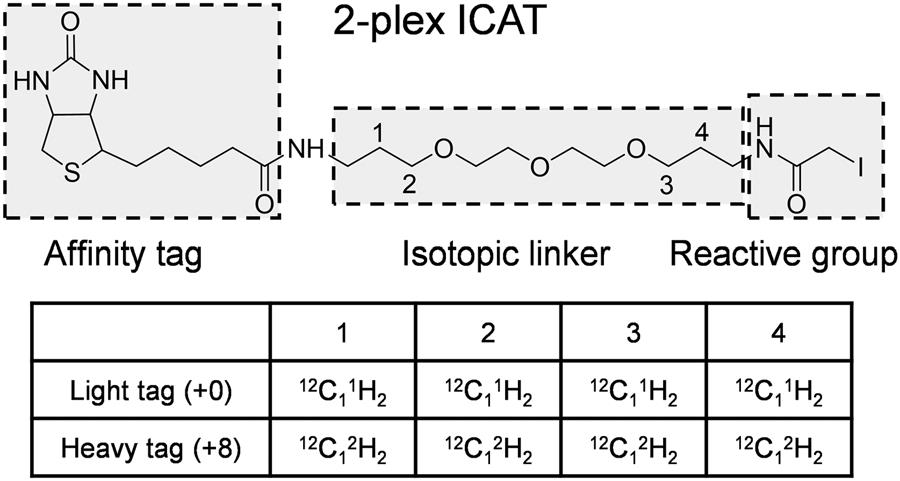
Figura 3 Diseño de estructuras y distribución de isótopos de ICAT 2 plex. ICAT, Isotope-Code Affinity Tag.
Este esquema en el diseño de la estructura de marcadores se ha mantenido en el desarrollo de nuevos ligandos marcados en los que se ha variado el tipo de grupo reactivo o el tipo de etiqueta. La mejora en el diseño de nuevas etiquetas surge de las principales limitaciones que se observaron con esta técnica.
La principal ventaja que ofrecía era la capacidad de cuantificar simultáneamente dos muestras en una única inyección LCMS y la reducción de la complejidad de la muestra, porque la etiqueta se dirige específicamente a las cisteínas, un aminoácido relativamente raro que constituye solo el 1,42% de todos los aminoácidos.
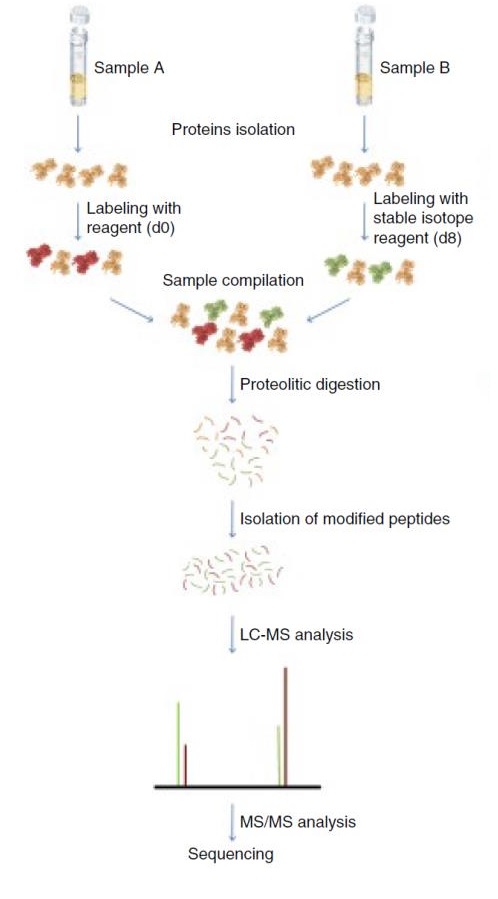
Figure 4 Esquema de experimento ICAT
Las principales limitaciones que surgieron se deben: al marcaje con 2H ya que alteraba los tiempos de retención de la cromatografía (la nueva versión de ICAT, introduce nueve átomos de 13C en lugar de ocho 2H eliminando así el efecto isotópico del 2H en la cromatografía); a la fragmentación de la biotina (afectando a la identificación del péptido); debido a la gran masa de reactivo ICAT, las m/z de los péptidos marcados pueden desplazarse fuera del intervalo de m/z de detección; y en último lugar, solo hay dos formas de marcas disponibles, lo que podría dar como resultado múltiples ensayos si es necesario comparar más de dos estados aumentando el costo del experimento.
La necesidad de realizar comparaciones de un mayor número de tratamientos llevó al desarrollo de técnicas de etiquetado donde se pueden comparar más de una muestras en un solo análisis (2-plex ICPL, 3-plex mTRAQ, 2-plexmTMT, 6-plex NeuCode SILAC, 3-plex DiPyrO tag).
CONCLUSIÓN
Todos los enfoques de cuantificación basados en MS1 tienen la fortaleza común de analizar simultáneamente múltiples muestras en una sola carrera de LC-MS, lo que mejora la precisión de la cuantificación. Sin embargo, la capacidad de multiplexación suele estar limitada por dos razones:
- El etiquetado isotópico multiplica la complejidad de los espectros de MS1 con el número de muestras etiquetadas. El aumento de la complejidad de MS1 es una desventaja cuando se usa DDA.
- Aumento de la capacidad de multiplexación. La limitación radica en el requisito de más átomos marcados isotópicamente y el desplazamiento de masa de al menos 4 uma entre etiquetados para evitar la superposición de las envolventes.
AUTOR
Gemma Barroso García, MSc.
Responsable SAI-Proteómica.
Hospital Nacional de Parapléjicos.
Toledo, España.
Referencias
- Tian X, Permentier HP, Bischoff R. Chemical isotope labeling for quantitative proteomics. Mass Spectrom Rev. (2021);1‐31. https://doi.org/10.1002/mas.21709
- Bachor R, et al; Trends in the design of isobaric labeling reagents for quantitative proteomics. Molecules 2019, 24, 701; doi:10.3390/molecules24040701 http://www.mdpi.com/journal/molecules
