Una de las técnicas más importantes y desafiantes en proteómica es la cuantificación de las diferencias entre dos o más estados fisiológicos de un sistema biológico.
La pregunta principal a la hora de enfrentarse a un experimento cuantitativo es saber qué método de todos los existentes es el más idóneo para nosotros, por eso en esta newsletter queremos presentar las diferentes opciones que existen.
Primero de todo debemos tener en cuenta que a pesar del increíble impacto de la espectrometría de masas y las técnicas de separación de péptidos en proteómica, la identificación y la cuantificación de todas las proteínas de un sistema biológico sigue siendo un desafío técnico no resuelto. La abundancia proteica y la complejidad de la muestra son factores significativos que afectan la disponibilidad de las proteínas para su cuantificación (Figura 1).
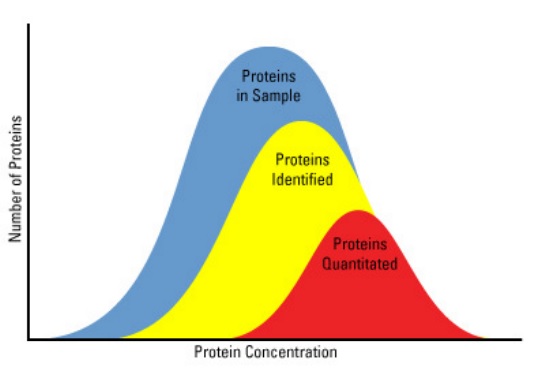
Figura 1. Representación esquemática de la fracción de un proteoma que puede identificarse o cuantificarse mediante espectrometría de masas. Las proteínas en las células abarcan una amplia gama de expresión y las tecnologías actuales basadas en espectrometría de masas suelen identificar solo una fracción de todas las proteínas presentes en una muestra. Debido a la limitada calidad de los datos, solo una fracción de todas las proteínas presentes en la muestra puede ser cuantificada de forma fiable.
Los péptidos proteolíticos exhiben una amplia gama de propiedades fisicoquímicas tales como tamaño, carga, hidrofobicidad, etc. que conducen a grandes diferencias en la respuesta por espectrometría de masas. Para una cuantificación precisa, por lo tanto, generalmente se requiere comparar cada péptido individual entre experimentos. En la mayoría de flujos de trabajo proteómicos, esto técnicamente se puede lograr de diferentes maneras (Figura 2).
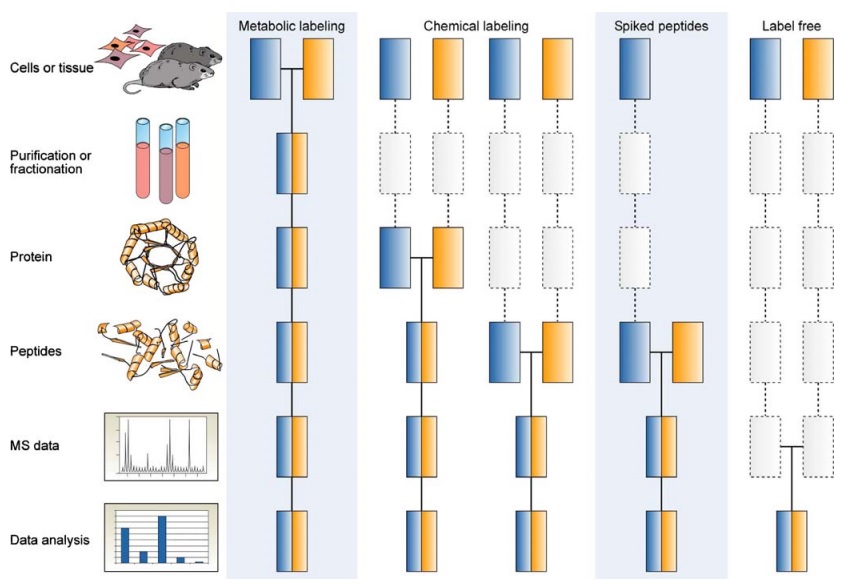
Figura 2. Flujos de trabajo comunes en espectrometría de masas cuantitativa. Las cajas en azul y naranja representan dos condiciones experimentales distintas. Las líneas horizontales indican el momento en que se combinan las muestras. Las líneas discontinuas indican puntos en los que la variación experimental y, por tanto, los errores de cuantificación pueden ocurrir.
Un enfoque importante se basa en la teoría de la dilución de isótopos estables que establece que un péptido marcado con isótopos es químicamente idéntico a su nativo y por lo tanto los dos péptidos también se comportan de manera idéntica durante la cromatografía y/o el análisis por espectrometría de masas. Dado que un espectrómetro puede reconocer la diferencia de masa entre el péptido marcado y el no marcado, la cuantificación se logra mediante comparación de sus respectivas intensidades de señal.
El marcaje con isótopos se puede introducir como un estándar en aminoácidos de estas cuatro maneras:
- Metabólicamente.
- Químicamente.
- Enzimáticamente.
- Con péptidos sintéticos como estándares externos.
Más recientemente, han surgido estrategias alternativas sin marcaje como las denominadas “label free quantitation” (LFQ). El objetivo de los métodos sin marcaje es comparar dos o más experimentos:
- Comparando el intensidad de la señal por espectrometría de masas directa para cualquier péptido.
- Utilizando el número de espectros adquiridos coincidentes con un péptido/proteína como indicador de su cantidades respectivas en una muestra dada.
Como puede contemplarse tanto en la Figura 3, como en la Tabla 1 cada método de cuantificación cuenta con fortalezas y debilidades que en próximas newsletter iremos desarrollando una a una más detalladamente.

Figura 3. Representación esquemática de enfoques proteómicos cuantitativos. A la izquierda, flujos de trabajo simplificados de tres enfoques “bottom-up” ampliamente utilizados donde se muestran el marcaje metabólico, el marcaje químico y la cuantificación sin marcaje o “label free”. A la derecha, un experimento típico de proteómica “top-down” utilizando 2D-DIGE como método de cuantificación. Las ventajas y desventajas de los enfoques particulares se resumen en cuadros rojo y azul, respectivamente. Abreviaturas utilizadas en esta figura: SILAC: marcaje de isótopos estables por aminoácidos en cultivo celular; iTRAQ: marcajes isobáricos para cuantificación relativa y absoluta; TMT: etiqueta de masa en tándem; 2D-DIGE: electroforesis diferencial en gel bidimensional.
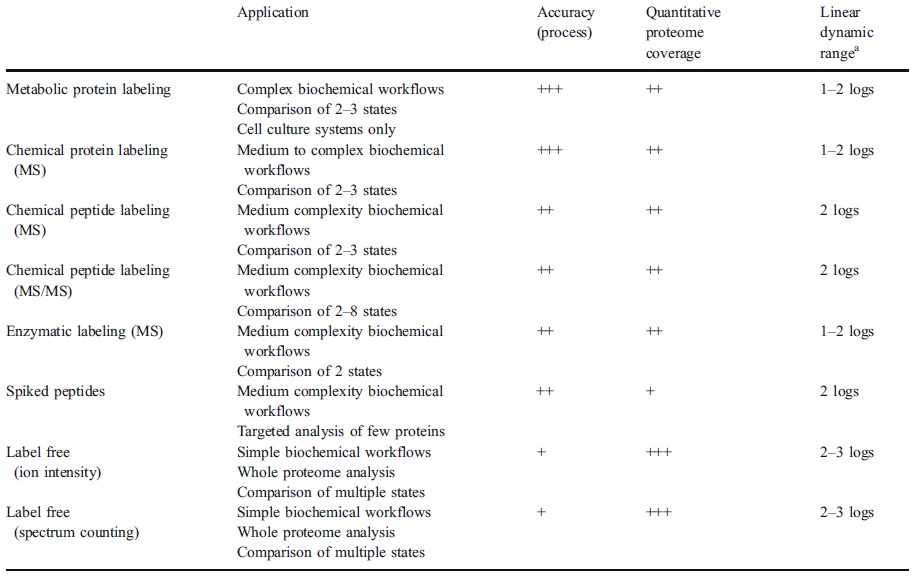
Tabla 1. Características y aplicaciones de los métodos de cuantificación por espectrometría de masas.
CONCLUSIÓN
Han surgido multitud de métodos a lo largo de los años para la cuantificación proteica de muestras simples y complejas utilizando la espectrometría de masas y cada vez se va refinando más tanto la parte metodológica como la estadística para el tratamiento de los datos obtenidos.
Y aunque aún estamos lejos de tener una cobertura total en la cuantificación, el desarrollo de las nuevas tecnologías junto con un diseño experimental adecuado y las estadísticas correctas puede llevarnos a la generación de buenos y concluyentes resultados.
En las siguientes newsletter iremos desarrollando más en detalle cada una de las técnicas cuantitativas más utilizadas en proteómica donde veremos su desarrollo y aplicabilidad.
AUTOR
Alba González Arandilla, MSc.
Especialista en espectrometría de masas.
Hospital Nacional de Parapléjicos.
Toledo, España.
REFERENCIAS
- Quantitative mass spectrometry in proteomics: a critical review. Anal Bioanal Chem (2007) 389: 1017–1031. DOI 10.1007/s00216-007-1486-6.
- Proteome Analyses of Hepatocellular Carcinoma Article in Journal of Clinical and Translational Hepatology. March 2014. DOI: 10.14218/JCTH.2013.00022
