El campo de la proteómica consta de una amplia gama de metodología, que ha sido impulsada en gran medida por el desarrollo moderno de la tecnología involucrada. El comienzo de la investigación proteómica se inició debido a desarrollos paralelos en cuatro áreas: (a) electroforesis en gel bidimensional (2D-PAGE), (b) métodos de espectrometría de masas, (c) bases de datos variadas con proteínas catalogadas, y (d) desarrollo de herramientas bioinformática. Hoy nos vamos a centrar en uno de los flujos de trabajo típicos en proteómica, “bottom-up proteomics”. En la revisión que realizan Emmalyn J. Dupree et al (1), presentan este enfoque desde una perspectiva de cromatografía líquida acoplada a espectrometría de masas, donde discuten su desarrollo multidisciplinario, lo bueno y lo malo, y el futuro con la mejora de la metodología actual.
BOTTOM-UP PROTEOMICS
Empecemos por recordar los pasos principales de los que consta este flujo de trabajo:
- Aislamiento de proteínas de la muestra a estudiar.
- Cuantificación de la concentración.
- Fraccionamiento de proteínas mediante electroforesis en gel o cromatografía líquida.
- Escisión enzimática (mediante tripsina generalmente) de las proteínas.
- Análisis de los péptidos generados por espectrometría de masas.
- Búsqueda en bases de datos para la identificación de proteínas.
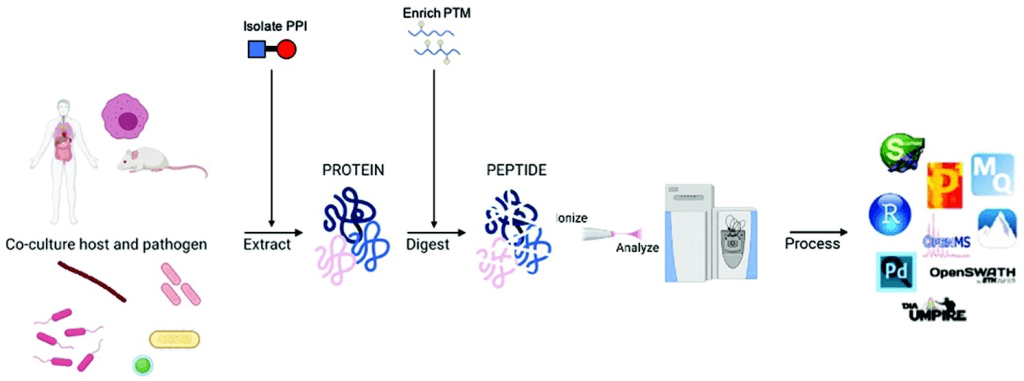
Fig. 1 Flujo de trabajo para “bottom-up proteomics” (Sukumaran A., et al 2021)
En este tipo de enfoque, también denominado “shotgun”, se utiliza las ventajas que los péptidos tienen sobre las proteínas: separación fácil mediante cromatografía, buena ionización y fragmentación predecible. Nos proporcionan grandes listados de identificaciones de proteínas y se utiliza para resolver la mayoría de los proteomas completos, complejos y disponibles en la actualidad.
Los tipos de adquisición que se utilizan en este tipo de estrategias son dependientes de datos (DDA).
No obstante, la eficiencia de las proteasas utilizadas y la limitación en el número de péptidos para la identificación de una secuencia no siempre es suficiente para identificar proteoformas. Aunque es posible que a través de la medición de cambios de masa, la identificación de isoformas y PTM sin conocimiento previo es extremadamente limitada en este tipo de aproximaciones.
Pasemos a revisar cada uno de los pasos y ver sus ventajas, inconvenientes y cuál puede ser su mejora en un futuro
PREPARACIÓN DE LA MUESTRA
Como hemos comentado en otras ocasiones, la preparación de la muestra es el primer y más importante paso. El principal cuello de botella ya que la sensibilidad y el rendimiento de los pasos posteriores están en juego.
A diferencia de otras “ómicas”, en proteómica no se dispone de un tampón universal para aislar proteínas. Los grupos funcionales de las cadenas laterales de aminoácidos permiten una diversidad tan grande en términos de carga proteica e hidrofobicidad que hace que las propiedades fisicoquímicas entre proteínas sean muy diversas y lo más importante, su variedad en abundancia.
Algunas proteínas se pueden encontrar en cantidades tan bajas como una proteína por célula, mientras que otras hasta varios millones por célula. A pesar de la gran selectividad y sensibilidad de los espectrómetros de masas, ningún instrumento disponible en la actualidad puede resolver tal complejidad de rango en una sola medición. Por lo tanto, se deben utilizar varias estrategias de fraccionamiento para mejorar la profundidad y cobertura del análisis proteómico.
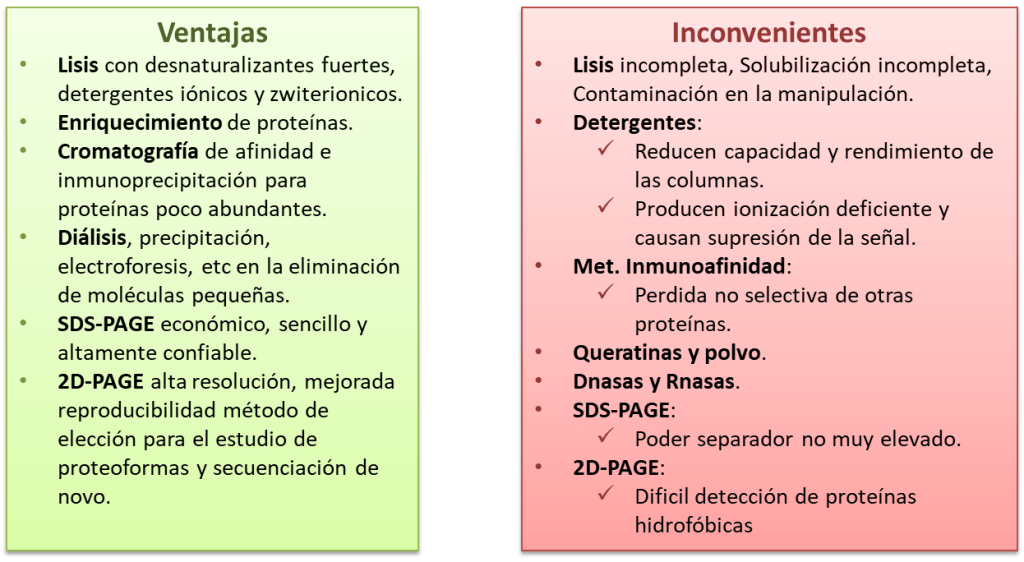
Idealmente, lo que se necesita es un método confiable y universal para extraer y fraccionar proteínas; uno que sea rápido, no requiera detergentes, requiera pasos mínimos y manipulación de muestras, y se pueda automatizar.

FRACCIONAMIENTO CROMATOGRÁFICO
El acoplamiento de la cromatografía líquida a los espectrómetros de masas inició el desarrollo de nuevos métodos de ionización y condujo a una diversidad de interfases destinadas a separar mezclas biológicas. Actualmente, es la técnica analítica común e indispensable para la investigación proteómica.

La metodología HPLC/UHPLC-MS seguirá siendo muy útil en las ciencias «ómicas» y se desarrollarán métodos de referencia, pero lo más deseable es la transición a diversas aplicaciones clínicas.

ANÁLISIS DE MS: INSTRUMENTACIÓN
No os voy a volver a recordar en que consiste un espectrómetro de masas (2), pero brevemente puntualizar que la electropulverización (ESI) es la técnica de ionización con la que mejor se acoplan los cromatógrafos de líquidos y que los analizadores son la clave para mantener la sensibilidad, la precisión y la resolución de masa, así como para generar espectros de iones ricos en información a partir de fragmentos de péptidos.
Dependiendo de la técnica de disociación para producir esos fragmentos se obtendrá más o menos información de la secuencia peptídica.
Tipos de disociaciones:
- CID: Disociación inducida por colisión. Es la más utilizada y común en la mayoría de las equipaciones.
- ECD: Disociación por captura de electrones.
- ETD: Disociación por transferencia de electrones.
- HCD: Disociación por colisión de alta energía


ANÁLISIS DE DATOS DE MS
El análisis de datos es un factor limitante en muchos experimentos proteómicos. Existen muchos softwares diferentes que facilitan y ayudan en el procesado de datos, sin embargo, estos programas también tienen limitaciones.
Para poder comprender los problemas en el análisis de datos, es importante comprender los conceptos básicos de fragmentación de péptidos. La mayoría de los problemas surgen de la falta de información de secuencia completa.
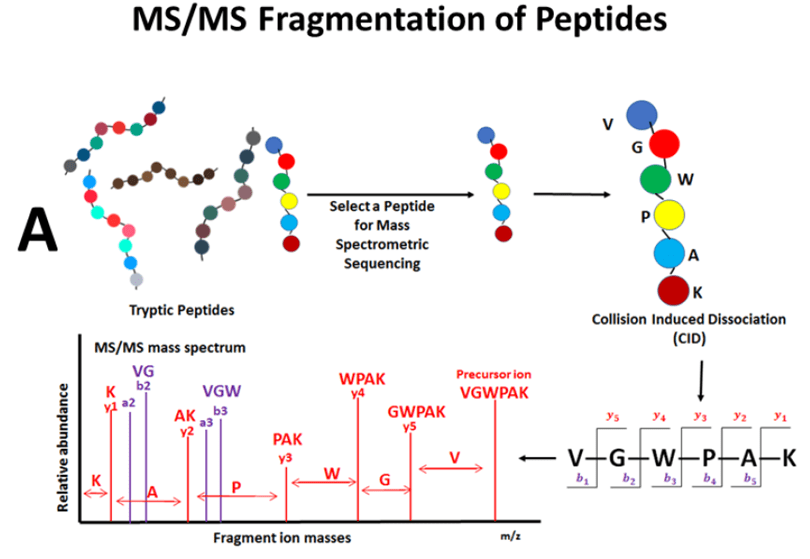
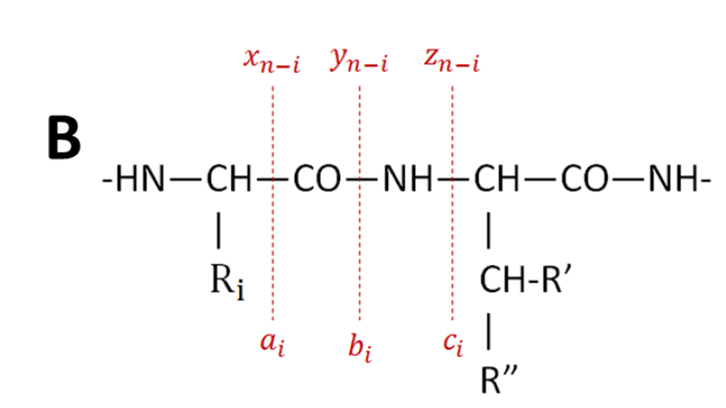
Fig. 2 Correspondencia entre la secuencia de aminoácidos del péptido y los picos de iones de fragmentos que se producen en MS/MS (A) y los principales iones fragmento teóricos que se producen a partir de la fragmentación de un péptido (B).
Uno de los principales problemas que debe abordarse en el análisis de datos proteómicos es la falta de bases de datos de proteínas bien anotadas. Es necesario que los investigadores ayuden a desarrollar estas bases de datos para que puedan utilizarse en futuros estudios de proteómica. Cuando se utiliza una base de datos de referencia para análisis proteómicos, se deben seguir parámetros de búsqueda estrictos y relevantes para disminuir la probabilidad de falsos positivos.
Siempre que sea posible, deben usarse bases de datos de proteínas pequeñas en lugar de grandes para minimizar los falsos positivos, los umbrales de péptidos deben establecerse en una tolerancia razonable y deben tenerse en cuenta las PTM relevantes. Además, si las identificaciones de péptidos son particularmente importantes, es aconsejable verificarlas utilizando los espectros de datos sin procesar.
FALSOS POSITIVOS, FALSOS NEGATIVOS Y ESPECTROS SIN ASIGNAR
Nuestro objetivo final es la identificación de proteínas presentes en la muestra de interés y su relevancia biológica. Este es un proceso de dos pasos que incluye encontrar PSM y concluir la identidad de las proteínas en función de las alineaciones de secuencias. Cada paso depende de una base de datos y, en cada paso, a medida que la base de datos crece, aumenta la probabilidad de encontrar péptidos y, por tanto, proteínas debido al azar. Los resultados que no están realmente presentes en la muestra se denominan identificaciones de falso positivo y solían ser el problema más desafiante de la proteómica.

Los resultados falsos positivos, los falsos negativos y los espectros no asignados solo pueden resolverse mediante avances en la búsqueda de bases de datos y análisis bioinformático, es decir, software adicional que permita la identificación de péptidos que corresponden a espectros MS/MS no asignados previamente.
CONCLUSIÓN
La MS y los métodos proteómicos han avanzado mucho en el campo del análisis de proteínas para la investigación fundamental y clínica. Existen métodos bien establecidos para la preparación de muestras y una excelente instrumentación para el análisis de MS. También hay una variedad diversa de motores de búsqueda de bases de datos. Sin embargo, el mayor desafío al que aún nos enfrentamos es una búsqueda completa y exhaustiva en bases de datos de un conjunto de antecedentes de proteómica. Está claro que lo que más necesita la proteómica es un soporte bioinformático mejor y más potente para realización de las búsquedas.
AUTOR
Gemma Barroso-García, MSc.
Responsable del Servicio de Proteómica.
Hospital Nacional de Parapléjicos.
Toledo, España.
Referencias
- Emmalyn J. Dupree et al(2020). A critical review of bottom-up proteomics: The good, the bad, and the future of this field.
- NL-PTMC04_Guía para principiantes.
- NL-PTMC01_SWATH-MS.
- PTMC: PostTranslational Modifications
- *SPEED: Sample Preparation by Easy Extraction and Digestion.
- iST: in-Stage Tip.
- Strap: Suspension Trapping.
- SP3: Single-Pot Solid-Phase-enhaced Sample Preparation.
- *PSM: peptide-spectrum match
