Las tecnologías de análisis de célula única están proporcionando un conocimiento sin precedentes sobre la complejidad de los sistemas biológicos. Las herramientas clásicas analizan poblaciones celulares en masa y, en consecuencia, una parte de los procesos biológicos permanece invisible. Un ejemplo es la existencia de ciertos tipos celulares que están muy poco representados (raros), pero que pueden tener importantes funciones fisiológicas o patológicas.
Con estas tecnologías, que se vienen desarrollando desde al año 2015, se está encontrando que las poblaciones celulares son más heterogéneas de lo que se había imaginado nunca. Cada célula única es diferente en términos de espacio (posición dentro de un tejido u órgano), tiempo (fase del ciclo celular, estado de activación, fase de desarrollo etc) y perfil molecular.
La citometría de flujo multiparamétrica compleja, que introdujimos en la anterior newsletter, nos permite llegar a un nivel de resolución de poblaciones celulares muy bueno, pudiendo identificar subpoblaciones celulares poco representadas o eventos raros. Además, está resultando una herramienta muy útil para diseccionar la complejidad fenotípica y funcional de las distintas poblaciones celulares de una muestra, tejido u órgano. Sin embargo, si queremos relacionarlas con los niveles de expresión génica o proteica tenemos que recurrir a otras tecnologías que nos permitan identificar el perfil de cada una de las células a nivel de RNA, DNA o de proteína.
Como sabemos, la citometría es toda aquella tecnología que permita medir características de las células. La citometría genómica es un conjunto de técnicas que nos van a permitir medir otras características como la expresión génica, pero a nivel de célula única, y además relacionarlas con otras características como la expresión de proteínas, de ahí que podamos considerar todas estas tecnologías como una aproximación multiómica.
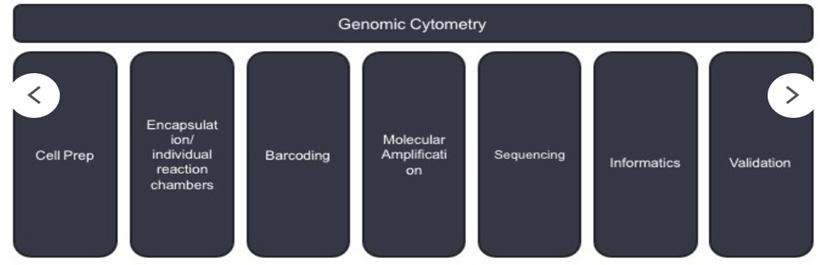
Fig.1: Workflow de experimentos de citometría genómica
Todas las tecnologías de análisis de célula única dependen de la separación física de las células de forma individual en un volumen de reacción que permita la aplicación de tecnologías posteriores de secuenciación de DNA o RNA o de análisis proteómico. Esto va a permitir caracterizar el genoma, el transcriptoma o el proteoma de cada célula individual.
Existen tres métodos principales de separación y preparación de las muestras para la realización de técnicas de análisis de célula única:
- Métodos de separación en placa multipocillo basados en citometría de flujo.
- Métodos basados en microfluídica o separación en nanopocillo.
- Métodos de indexado combinatorial basados en citometría de flujo.
Aunque también existen métodos basados en imagen y métodos de transcriptómica espacial, no son objetivo de este artículo (ver referencia Salomon R et al 2020).
A continuación, pasamos a explicar un poco más en detalle cada uno de ellos
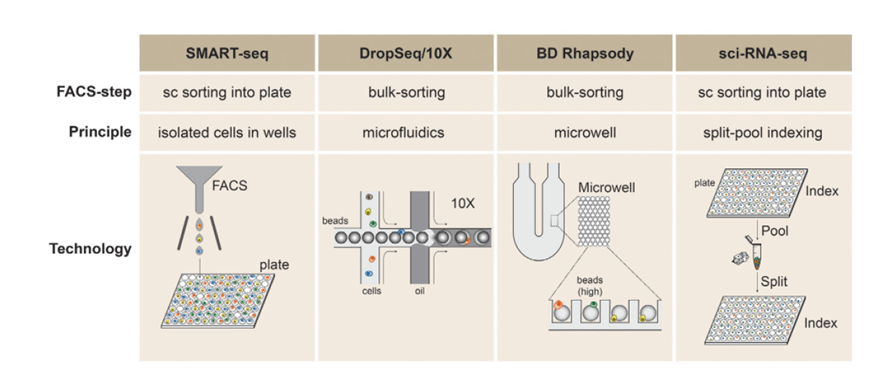
Fig. 2: Métodos para la separación y preparación de muestras para tecnologías de análisis de célula única
Separación de células individuales en placas multipocillo por citometría de flujo
Es el método clásico utilizado para la obtención de células únicas. Se basa en la separación celular activada por fluorescencia (FACS= “Fluorescence-Activated Cell Sorting”) de las células de interés en base a unos parámetros de fluorescencia determinados. De esta forma, lo que se hace es definir una población celular determinada y se programa al citómetro separador para depositar una única célula en cada pocillo de una placa, ya sea de 96 pocillos o de 384, en los cuales previamente hemos añadido el volumen de tampón de reacción correspondiente según la técnica posterior de análisis que queramos realizar.
Por ejemplo, en el caso de querer obtener información sobre el transcriptoma de una célula se podrá utilizar un sistema como el Smart-seq2 que permite hacer una primera reacción de retro-transcripción con un material de partida de incluso 50 pg de RNA, y una posterior amplificación de cDNA para la obtención de una librería que se secuencie posteriormente.
Separación de células por microfluídica o en nanopocillos
Son sistemas que incrementan la capacidad de analizar un mayor nº de células y que eliminan ciertos errores o imprecisiones de pipeteos para realizar las reacciones posteriores a la separación celular.
Existen dos sistemas: uno que utiliza microchips en los cuales se van a ir combinando células individuales con esferas marcadas con un código de barras en forma de oligonucleótido en el interior de gotas de reacción, y un segundo método que también deposita células individuales junto con esferas marcadas con oligonucleótidos, pero lo hace en nanopocillos impresos en un microchip dónde se produce la reacción posterior de amplificación de RNAs y generación de librerías como en el caso anterior.
Su ventaja principal es que las esferas que se combinan con las células tienen todos los reactivos necesarios para la amplificación y generación de librerías, evitando así la necesidad de preparar la reacción a posteriori (implica menos errores o imprecisiones).
Indexado combinatorial basado en citometría de flujo
Este sistema se basa en una primera separación de las células de forma individual, cada una en un pocillo mediante FACS, y la adición de un primer código de barras a cada uno de los pocillos. A continuación, se hace un pool con todas las células o núcleos separados y se realiza una segunda separación en placa, añadiendo un nuevo código de barras a cada pocillo. De esta forma, cada una de las células posee un código único de identificación por la combinación de varios códigos de barras.
El tratamiento posterior de las muestras viene siendo el mismo que lo expuesto anteriormente.
MULTIÓMICA
Aunque la transcriptómica es una fuente de información muy importante, existen otras lecturas que pueden revelar otras diferencias entre unas células u otras. Así, se podrían hacer análisis multiómicos: RNA con DNA, RNA con proteína, DNA con proteína o incluso RNA con modificaciones epigenéticas (metilaciones etc). Las aproximaciones metabolómicas y proteómicas están comenzando a desarrollarse poco a poco, pero serán importantes en un futuro próximo.
Actualmente una de las aproximaciones multiómicas que se pueden realizar es relacionar los datos de transcriptómica obtenidos con la expresión de proteínas analizada por citometría de flujo.
Cuando realizamos la tinción de las células con anticuerpos o sondas que caracterizan distintas poblaciones celulares, vamos a obtener datos en masa de las poblaciones en su conjunto. Sin embargo, al realizar la separación por FACS podemos pedirle al citómetro que guarde los datos de fluorescencia y dispersión de la luz para cada una de las células que está depositando en cada pocillo. Este proceso se denomina Index Sorting.
Además del Index Sorting existen otros métodos para poder relacionar los datos obtenidos de la transcriptómica con la expresión de proteínas, como puede ser CITE-seq. CITE-seq utiliza anticuerpos marcados con oligonucleótidos específicos que convierten la detección de proteínas en una secuencia de DNA que puede ser cuantificada. Así, los oligos unidos a los anticuerpos actúan como transcritos sintéticos que van a ser retrotranscritos, amplificados y secuenciados simultáneamente con los transcritos de la célula en cuestión. De esta forma, cuando se obtienen los datos de los mRNAs expresados por cada una de las células, se obtienen también los datos de qué proteínas están expresando e incluso sus niveles de expresión.
La relación de unos parámetros con otros (expresión protéica – RNA) requerirá un análisis de los datos diferente al análisis de datos de citometría convencional, que incluye técnicas de reducción dimensional y clustering, similares a las que se pueden usar para el análisis multiparámetrico complejo, y que veremos en próximas newsletters.

Fig.3: Workflow del análisis de datos
Lecturas recomendadas:
– Salomon R et al (2020) Cytometry A: https://doi.org/10.1002/cyto.a.24209
– Luecken M.D. and Theis F.J. (2019) Mol Sys Biol: https://doi.org/10.15252/msb.20188746
– Cossariza A et al (2019) Eur. J. Immunol: https://doi.org/10.1002/eji.201970107
