Algo en lo que insistimos en nuestros cursos de formación y que muchos alumnos nos preguntan es por la preparación de las muestras.
Los más veteranos ya sabréis la respuesta. No, no existe un protocolo perfecto o universal que funcione bien para todo tipo de muestras.
Las condiciones óptimas de preparación deben ser determinadas empíricamente para cada tipo de muestra. Dependiendo de la naturaleza de la muestra (tejido, fluido biológico, células), de la información que queramos obtener y de las técnicas que vayamos a utilizar para resolver nuestro problema biológico, adaptaremos unos métodos de separación, homogeneización, solubilización, limpieza, etc.
No he descubierto la pólvora ¿verdad?
Pues algo tan sencillo y que todos tenemos en nuestras rutinas, es el principal cuello de botella cuando se quiere realizar un análisis en proteómica. Elegir los métodos con los que obtengamos unos mejores rendimientos de extracción de proteínas con tampones compatibles a los protocolos de digestión y su posterior análisis por espectrometría de masas es fundamental para la generación de datos precisos e informativos.
Hoy en día, las tecnologías de última generación en proteómica permiten la identificación de miles de proteínas a partir de una pequeña cantidad de muestra, por ejemplo, clasificadas por FACS, microdiseccionadas con láser o células fijadas con formalina. Estos avances incluyen métodos mejorados de preparación de muestras, tecnologías de separación para llegar a una aproximación de proteómica unicelular basada en espectrometría de masas (MS). En la revisión de Lombard-Banek C. et al; 2020; podréis encontrar como los enfoques proteómicos se aplican en procesos de terapia celular.
Recordemos como es el proceso de trabajo en proteómica
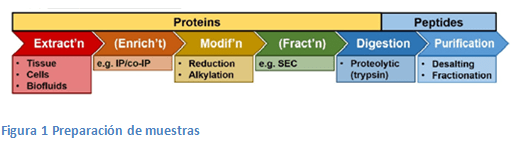
Primero, las muestras biológicas se preparan a través de un flujo de trabajo ascendente, en el que las proteínas se extraen de las células y se digieren enzimáticamente en péptidos. Los péptidos posteriormente se analizaran por LC-MS/MS.
Las aproximaciones que han surgido en los últimos años en el tratamiento de muestras tienden a que en un único vial se pueda realizar la gran mayoría de los pasos para aumentar el rendimiento del proceso, sobre todo cuando se parte de cantidades mínimas de muestra.
En la tabla se resume algunos de estos procedimientos y se comparan sus características.

FASP: Filter-aided simple preparation. La lisis de proteínas y la limpieza de péptidos se realizan en receptáculos separados en FASP, creando una fuente potencial de pérdidas de adsorción.
Pioneros en la mayoría de los avances en la preparación de muestras basadas en filtros generalmente usa filtros de corte de peso molecular (MWCO) que van desde 1 a 100 kDa de tamaño. La digestión enzimática se realiza en los propios filtros y los péptidos resultantes se liberan por centrifugación.
Ventajas: FASP mejoró las tasas de identificación de péptidos en comparación con el enfoque tradicional de digestión “in-solution”.
Inconveniente: FASP es un protocolo largo y requiere cantidades sustanciales de material.
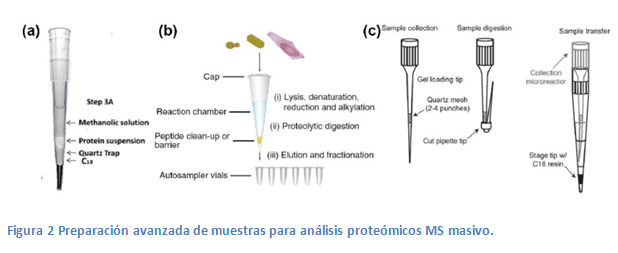
S-Trap: Suspensión trapping. Permite la limpieza, digestión y fraccionamiento de péptidos dentro de un solo dispositivo (Figura 2a). El dispositivo S-Trap está diseñado para un flujo de trabajo basado en la centrifugación y está compuesto por un filtro de cuarzo para atrapar proteínas intactas seguido de un tapón monolítico C18 (fase reversa) en el que se retienen los péptidos y se realiza la desalinización y / o fraccionamiento de la muestra.
Ventajas: El proceso en S-Trap se simplifica y es más rápido en comparación con el protocolo FASP debido a su diseño integrado. Se pueden preparar muestras con menos material proteico y el enfoque es compatible con una amplia gama de detergentes.
iST: In-Stage tip. Las células se pueden lisar directamente en el dispositivo (Figura 2b). El iST es simplemente una punta de pipeta con un inserto de disco C18 que sirve como barrera y dispositivo de limpieza de péptidos. Gracias al diseño de la punta, la preparación de múltiples muestras puede paralelizarse con el uso de pipetas multicanal o incluso automatizarse mediante dispositivos robóticos. De hecho, todo el procesamiento puede realizarse en un dispositivo de 96 pocillos, mejorando enormemente el rendimiento de la preparación de muestras.
Ventajas: En un estudio comparativo de iST con FASP, iST condujo a una mejor cobertura de proteínas de membrana y nucleares que FASP.
Inconveniente: iST no puede eliminar detergentes iónicos como SDS. Además, la muestra se calienta a alta temperatura antes de la digestión (> 60 ºC), por lo que debemos evitar agentes caotropicos como la urea.
Streamlined iST: Diseño combinado de S-Trap e iST. Para análisis proteómicos de células clasificadas por FACs (Myers, S.A. et al; 2019). Las células se recogen durante la clasificación directamente en la punta con el filtro de cuarzo quedando retenidas. La punta del filtro se dobla para evitar pérdidas durante la digestión y el pliegue se mantiene mediante un anillo cortado de una punta de pipeta. Las células se lisan y la proteína extraída se digiere (Figura 2c, en el centro). Luego se inserta la punta de preparación en una segunda punta que contiene un disco de C18 para la limpieza de péptidos (StageTip).
Ventajas: Reducción de la cantidad de partida de muestra, menos de 2 µg de contenido de proteína, lo que representa una disminución de ~ 5 a 100 en comparación con los enfoques tradicionales de EM (Tabla 1).
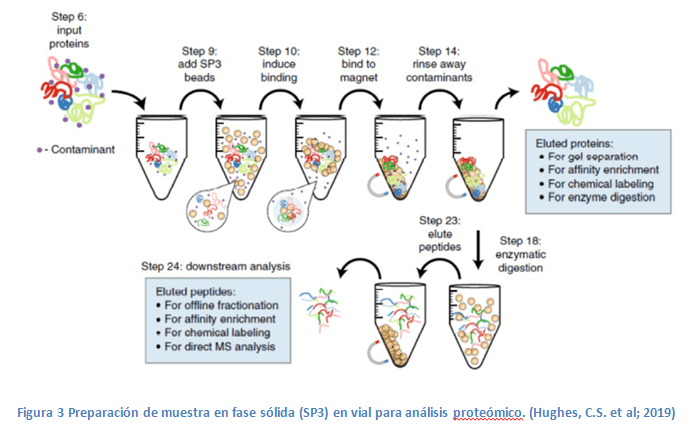
SP3: Single pot solid phase-enhanced simple preparation. Basado en el enfoque clásico de preparación de muestra en vial. Se utilizan bolitas mágneticas derivatizadas con grupos funcionales hidrofílicos (Figura 3). Este enfoque permite el enriquecimiento de proteínas, la eliminación de detergente y la digestión disolución al agregar las bolitas directamente en el lisado celular.
Ventajas: Todos los pasos del procesado de la muestra se realizan en un solo tubo y el costo del ensayo es menor que el de los enfoques descritos anteriormente. Este enfoque es compatible con una amplia gama de detergentes y agentes caotrópicos. Además, es compatible con un amplio intervalo de cantidad de partida de muestra, de muy baja a alta, al cambiar únicamente el tamaño del tubo de microcentrífuga utilizado y la cantidad de perlas magnéticas añadidas.
Bibliografía:
- Lombard-Banek C, Schiel JE. Mass Spectrometry Advances and Perspectives for the Characterization of Emerging Adoptive Cell Therapies. Molecules. 2020; 25(6):1396. Published 2020 Mar 19. doi:10.3390/molecules25061396 https://pubmed.ncbi.nlm.nih.gov/32204371/
- Sielaff M, Kuharev J, Bohn T, et al. Evaluation of FASP, SP3, and iST Protocols for Proteomic Sample Preparation in the Low Microgram Range. J Proteome Res. 2017; 16(11):4060‐4072. doi:10.1021/acs.jproteome.7b00433
- Pieragostino D, Lanuti P, Cicalini I, et al. Proteomics characterization of extracellular vesicles sorted by flow cytometry reveals a disease-specific molecular cross-talk from cerebrospinal fluid and tears in multiple sclerosis. J Proteomics. 2019;204:103403. doi:10.1016/j.jprot.2019.103403
- M. Rezaa Mohammadi, Milad Riazifar, Egest J. Pone, Ashish Yeri, Kendall Van Keuren-Jensen, Cecilia Lässer, Jan Lotvall, Weian Zhao, Isolation and characterization of microvesicles from mesenchymal stem cells, Methods, Volume 177, 2020, Pages 50-57, ISSN 1046-2023, https://doi.org/10.1016/j.ymeth.2019.10.010
